There are clusters of ER-shapingproteins
Modified ubiquitination assays on full-length GST-FAM 134B substrates for the diagnosis of multiple sclerosis
The modified ubiquitination assays were based on AMFR-mediated ubiquitination. The purified substrates were full-length GST–FAM134B. His name was RHD90 or 1 M. His–Ub–RHD90–264–Ub–Strept-II) was mixed with 10 µM Ub, 10 mM ATP and 10 mM MgCl2 in 50 mM Tris-HCl pH 7.5, 150 mM It takes 2 h at 37 C for NaCl, 100 nM E1 UBA1; and 0.8 M E2 of AMFR UBE2G2 to be absorbed. The reaction mixture was analysed using a western blotting method against His6 or Ub. The samples were prepared to be used for diagnosis of multiplesclerosis. The samples were put into a buffers consisting of 1%sodium deoxycholate and 1.5 mM TCEP. chloroacetamide and 50 mM heated for 30 minutes at 60 C. We then added 500 ng trypsin to each sample and incubated overnight at 37 °C. The reaction was stopped with 1% TFA in isopropanol. The drugs were cleaned up with stage tips. After one wash with 1% TFA in isopropanol and one wash with 0.2% TFA in water, peptides were eluted in 80% acetonitrile plus 1.25% ammonia. The dried and processed peptides were used in LC–MS.
ARL6ip1 Coarse-grained MD Simulation using AlphaFold and Grassmann Clustering Algorithms
ARL6ip1 was built using the AlphaFold program. Five models were constructed, and the top-ranked model was chosen as it had maximal overlap with predicted secondary structures and consensus transmembrane topology, a higher pLDDT score and a relatively lower predicted alignment error (AF confidence measure).
Coarse-grained (CG) MD simulations were prepared using the MARTINI model (v2.2)34,35. Initial CG structures were built using martinize.py36. A program called theDictionary of Secondary Structures inProteins was used to generate backbone Restraints. CG models were embedded into POPC (16:0–18:1 phosphatidylcholine (PC)) bilayers spanning the periodic simulation box in the x–y plane. The initial configurations for each system were solvated using a 150 mM pool of water. NaCl using insane.py36. The system was energy minimized and equilibrated by using the barostat 40 and the Berendsen thermostat and the production runs had a 20-fs time step. System temperature and pressure during the production phase were maintained at 310 K (unless otherwise stated) and 1 atm with the velocity rescaling thermostat41 and the semi-isotropic Parrinello–Rahman42 barostat, respectively. The simulations were done using gromacs. Long-lived, highly populated RHD’s were obtained by clustering evenly sampled conformations from each trajectory. The Gromos method45 was implemented in thegmx_cluster tool to obtain clusters.
Western blot data analysis using ImageJ software (version 1.51w) for protein assays in Escherichia coli
Quantifications of western blot signals were performed using ImageJ software (version 1.51w). For each assay, protein bands were quantified from at least three independent experiments. Data analysis was performed using Microsoft Excel 2016 (Microsoft Corporation) or Prism 9.4.1 (GraphPad Software). The mean standard deviation was graphed. The significance was determined by a single t-test. P > 0.05 or P ≤ 0.05 were regarded as not statistically significant or statistically significant, respectively. Two independent variables were included in the one-way analysis of the variance. Bonferroni’s multiple comparison test was performed.
Alternatively, membrane shaping by full-length GST–FAM134B ubiquitinated with AMFR was investigated by transmission electron microscopy using freeze-fractured liposomes prepared from Folch-fraction type I lipids (Sigma-Aldrich) as previously described25. The liposomes were put in the HN-buffer (20 mM HEPES/KOH pH 7.4, 150 mM NaCL and 2.5 mM DTT) for 15 minutes at 37 C. Subsequently, 15 μg proteinase K was added and incubated for 40 min at 45 °C to avoid liposomal aggregates26. The small aliquots of the suspension were then frozen. The samples were examined by systematic grid exploration using an EM 900 electron microscope (Zeiss) at 80 kV. Images were acquired using a wide-angle dual-speed 2K CCD camera (Tröndle). Diameters of liposomes were determined using ImageJ software (version 1.51w).
Trx–His fusion proteins were purified from Escherichia coli treated with 0.5 mM In the night, 18 C. Eluted proteins were concentrated using Amicon Ultra-4-10k centrifugal filter units (Millipore) and then either dialysed at 4 °C against HN buffer (20 mM HEPES/KOH pH 7.4, 150 mM NaCl and 2.5 mM dithiothreitol) (material used for Fig. 4 and Extended Data Fig. 5 analyses) or against liposome buffer (25 mM HEPES-KOH, pH 7.2, 25 mM KCl, 2.5 mM magnesium acetate and 100 mM potassium glutamate) (protein used for studies presented in Fig. 2).
We followed a protocol for protecting against proteases. The70,000 cells per well were seeding on 18mm coverslips covered in 0.1 quality line retarding (PLL) Cells were transfected with different constructs on the next day. Cells were washed after two days with a pre-warmed buffer. The buffer was put in a chamber filled with the same substance, and transferred to it. After a pre-permeabilization image is taken, 18 M digitonin is administered on the Cell Observer Z1, followed by a frame every 20 s. After 120 s, the buffer was replaced with a buffer with 6 mM of trypsin. To do the analysis, an outline of a cell was drawn and a fluorescent intensity was measured by taking the background intensity from a cell free spot. The area under the curve was used to calculate the area under the curve.
Proximity ligation assays (PLAs) were performed using a Duolink in situ red starter kit mouse/rabbit (DUO92101, Sigma-Aldrich) according to the manufacturer’s instructions with rabbit anti-FAM134B (ref. 1) and rabbit anti-ARL6IP1 (PRS3305, Sigma-Aldrich) antibodies using the Minus (DUO92010) and the Plus probe (DUO92009). Rabbit anti-FAM134B-Plus was used in a 1:5 dilution and rabbit anti-ARL6IP1-Minus in a 1:10 dilution on WT, Arl6ip1 and Fam134b KO MEFs after PFA (4%) fixation and permeabilization with 0.25% (v/v) Triton X-100 in PBS.
The MaxQuant-Perseus data processing and analysis of the ProteomeXchange Consortium32 (PXD032721) dataset
MaxQuant was used to process theMS raw data. Acquired spectra were searched against the human ‘one sequence per gene’ database (Taxonomy ID 9606) downloaded from UniProt (12 March 2020; 20,531 sequences), and a collection of 244 common contaminants (‘contaminants.fasta’ provided with MaxQuant v1.6.17.0) using the Andromeda search engine integrated into MaxQuant v1.6.17.0 (ref. 28,29). Identifications were filtered to obtain false discovery rates below 1% for both peptide spectrum matches (minimum length of seven amino acids) and proteins using a target–decoy strategy30. Protein quantification and data normalization relied on the MaxLFQ algorithm implemented in MaxQuant v1.6.17.0 (ref. 31). The ProteomeXchange Consortium32 has deposited the data with the dataset of the name PXD032721, in the PRIDE partner repository 33. 8b), PXD032741 (Fig. 4d and Extended Data Fig. 8c–e, and the extended data fig. 9h–k. PXD039186 (Extended Data Fig. 10m,n), PXD039187 (Fig. 5c) and PXD039188 (Extended Data Fig. 10p,q). The Uniprot human database included a list of common contaminants. The searches were conducted with tryptic specifications and default settings. Fixed modifications were defined to include methionine oxidation, and N-terminal acetylation. The match-between-run feature was used with a time window of 1 min. For further analysis, Perseus v2.0.7.0 was used and first filtered for contaminants and reverse entries as well as proteins that were only identified by a modified peptide.
Dried peptides were reconstituted in 2% acetonitrile, 0.1% TFA and analysed on a Q Exactive HF mass spectrometer coupled to an easy nLC 1200 (ThermoFisher Scientific) using a 35-cm-long, 75 µm inner diameter fused-silica column packed in-house with 1.9 µm C18 particles (Reprosil pur, Dr. Maisch) and kept at 50 °C using an integrated column oven (Sonation). While eluting the drugs, they were sprayed into the mass analyzer using a nanoFlex ion source. A maximum injection time of 20 ms, an automatic gain control target value of three charges, and full scans in profile mode yielded results at a resolution of 60,000 at m/z 200. Up to 15 of the most intense peptides per full Scan can be isolated and fragmented using higher energy collisional dissociation. MS/MS spectra were acquired in centroid mode with a resolution of 30,000, a maximum injection time of 45 ms and an automatic gain control target value of 1 × 105. Dynamic exclusion was set to 20 s to minimize the acquisition ofFragment spectrum of already acquired precursors that were single charged ion and unassigned charge state.
Source: https://www.nature.com/articles/s41586-023-06090-9
ARL6ip1 is a Candidate in Bulk Autophagy and Transfections of U2OS MEFs and Human Fibroblasts
MEFs or human fibroblasts were seeded in 6-well-plates and cultured to 70–80% confluency. Cells were washed with PBS, then put into new medium with a combination of 1.5 M Sigma and 5 g tunicamycin. After 24 h for MEFs and 48 h for human fibroblasts, the culture medium was removed, cells washed with PBS and trypsinized. All the cells were pooled and cultured at the same time. for 5 min (Heraeus Sepatech Megafuge 2.0R) and resuspended in fresh medium. Cell viability was measured by trypan blue exclusion with an automatic counting device (Bio-Rad TC20 automatic cell counter).
Arl6ip1 WT and KO MEFs were seeded in 24-well-plates at 40,000 cells per well. After 24 h, cells were transfected with the mCherry–GFP–FAM134B plasmid in combination with either the ARL6IP–HA or the ARL6IP1-7KR–HA plasmid. After 48h, cells were fixed with ice- cold methanol for 10 min, then permeabilized with 0.25% Triton X-100 in PBS and then blocked with 4% NGS for 1 h and stained for LC3B and HA. Images were taken with a LSM 880 and analysed using the ComDet (v.0.5.5) plugin for ImageJ (https://github.com/ekatrukha/ComDet; settings: particle size = 10 pixels, co-localization distance = 10, intensity threshold = 200/200/15). There were only signals that were analysed.
We knocked down ARL6ip1 in U2OS cells in order to assess whether it is involved in bulk autophagy. 6 h of 250 nM Torin1 exposure triggered the autophagy. The images were captured with a high-content microscope. To assess whether ARL6IP1 is involved in mitophagy, ARL6IP1, CRISPR–Cas9 KO HeLa cells were transfected with the mitophagy reporter mCherry–GFP–FIS1. Autophagic flux was triggered with 40 μM CCCP for 4 h. Pexophagy was assessed shortly after the introduction of the mCherry–GFP–PMP34 reporter in U2OS cells. It was possible to acquire images with a confocal microscope. The red and yellow puncta were counted on their own.
U2OS cells were put in Ibidi -slide VI chambers at 70% confluency. The cells were fixed for 30 min with pre-warmed (37 °C) 4% methanol-free formaldehyde (Sigma-Aldrich) in PBS followed by three washes with PBS. Fixed cells were then incubated in permeabilization/blocking buffer (10% FBS and 0.1% saponin) containing primary antibodies for 60 min at room temperature: rabbit anti-FAM134B (Genscript), mouse anti-LC3B or mouse anti-REEP5, each diluted 1:200. Excess primary antibodies were removed from the chambers by three washes with PBS. After 60 minutes in the permeabilization/blocking buffer cells were removed from the sample with three washes with PBS. The sample were washed with PBS at room temperature for 10 min, and then post-fixed with 4% MHFA in PBS. 125-Nanopartz gold beads were used in the exchange DNA-PAINT experiments. The gold beads were sonicated twice, once in PBS and once again in 10 min. We added 100 l of gold bead solution to each slide and washed it three times. The cell chambers were finally connected to a microfluidic device (Bruker). Before imaging, ATTO-655-labelled imager strands R1 or R2 (50 pM, 0.5 M NaCl and PBS, pH 8.3) were injected into the flow chamber at a flow rate of 200 µl per minute. In the exchange DNA-PAINT experiments, imager strands were exchanged between sequential imaging cycles by washing the samples with PBS and injecting new imager strands under equal conditions.
RNA was isolated by TRIzol–chloroform extraction. RNA was reverse-transcribed using a GoScript reverse transcription kit (Promega). A final 20 ng of cDNA and EvaGreen mix was used for a final calculation of the result.
Detection of ARL6ip1 and its associated fibroblasts by means of membrane-activated PVDF membranes in mice, mouse and human
Proteins were denatured at 90 °C for 5 min in Laemmli buffer, resolved by SDS–PAGE and transferred to methanol-activated PVDF membranes (Amersham Hybond P 0.45 µm). In 10% skim milk, the Membranes were blocked for 1 h. Tris, 150 mM NaCl, 0.1% Tween 20, pH 7.5) and incubated overnight at 4 °C with the specific primary antibody followed by 1 h incubation with the respective secondary antibody at room temperature. Detection was carried out using Clarity Western ECL substrate (Bio-Rad) and a LAS 4000 automated detection system (GE Healthcare). Bands were quantified using ImageJ.
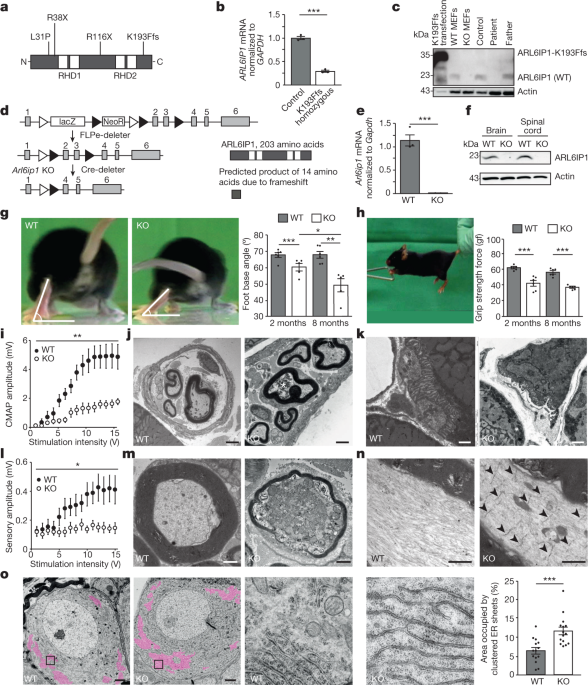
We studied the function and consequences of ARL6ip1 in mice and different cell culture models, as well as the fibroblasts of a patient who had the homozygous mutation AR6ip1K193Ffs. Mouse experiments were performed on a C57BL/6 background after backcrossing for more than four generations. Groups of up to 3 mice per cage were maintained with maximum temperatures between 21 2 C and 500 lx. Mice had free access to standard mouse chow and water. Experimental groups were assigned to the littermates of the same sex. Experiments were conducted in a blinded manner with regard to cell, mouse and human genotypes. The replicate experiments used to generate the datasets are included in the figure legends. All animal experiments were approved by the Thüringer Landesamt für Lebensmittelsicherheit und Verbraucherschutz (registration numbers 02-055/14 and UKJ-17-006). Studies using human fibroblasts were approved by the local ethics committee.
Introduction and Sequencing of Vector cDNAs into Vector pDONR223 and Subcloning into the Vector GEX-6P1 Using the BP Clonase Reaction Kit
We introduced the cDNAs into vector pDONR223 using the BP Clonase Reaction Kit (Invitrogen) followed by transfer to GATEWAY destination vectors using the LR Clonase Reaction Kit (Invitrogen), resulting in vectors pLTD-N-HA-PURO, pcDNA5-FRT/TO-N-mCherry-eGFP, pcDNA3.1-N-HA, pMH-SFB, pGEX6P1-DEST, pDEST 527 6×His, pDEST-V1ORF, pDEST-V2-ORF, pDEST-ORF-V1 and pDEST-ORF-V2. GST–FAM134B was generated by subcloning into the vector pGEX-6P1 using the EcoRI site. The EcoRV site and SmaI site were targeted by the GST–FAM134B 17KR.
Primers are presented in Supplementary Table 2, primary antibodies in Supplementary Table 3 and secondary antibodies in Supplementary Table 4.
The donor blastocysts had the EUCOMM embryo stem cell clone injected into them. Next, 15–30-week-old F1 female offspring from C57BL/6J and CBA/J matings served as foster mice. The chimeras were bred with C57BL/6J. For all experiments, littermates were used, which had been backcrossed for at least four generations. Arl 6ip1 FORWARD, Arl6ip1-KO-reverse and Arl6ip1 COMMENTS were used in the genotyping.
The U2OS TRex cell lines were used to make stable cell lines. For ARL6IP1 knockdown experiments, the respective cells were transfected with either 30 pmol siNT (non-targeting sequence, Qiagen) or with 30 pmol double-stranded ARL6IP1 siRNA (Integrated DNA Technologies; hs.RiARL6IP1.13.2) using Lipofectamine RNAiMAX transfection reagent (Invitrogen, 13778075). For ARL6IP1 and FAM134B double-knockdown experiments, HeLa cells were transfected with ARL6IP1 siRNA and FAM134B siRNA (siRNA RETREG1 18 J-016936-18-0002 and siRNA RETREG1 21 J-016936-21-0002, respectively). Experiments were performed 72 h after transfection.
Source: https://www.nature.com/articles/s41586-023-06090-9
Microscopy and electrophysiology of peripheral nerves of anaesthetized mice on elevated beams: Effects of stimulus intensity for the walking test and motor fibre analysis
For the beam walk test, mice were placed on an elevated beam of 1 m in length and 4 cm in width, with the home cage at the end. After habituation on three consecutive days, the mouse was videotaped from behind during its movement on the beam. When the toe was lifted, the hind limb’s foot base angle was measured.
For the grip strength analysis, mice were lifted at the tail base, brought to a trapeze-shaped handle connected with a force sensor (Grip Strength Meter, Ugo Basile). The mouse was slowly pulled away from the handle after it was released when it spontaneously grabbed it.
For the electrophysiological analysis of peripheral nerves, anaesthetized mice (100 mg kg–1 ketamine and 16 mg kg–1 xylazine) were placed on a heating pad. The first pair of needles were inserted near the base of the tail with the second pair 30mm away from the stimulation site. For the analysis of motor fibres, the stimulus was applied through the proximal electrodes and the response recorded using the distal electrodes. Compound muscle action potentials and sensory nerve action potentials were evoked with increasing intensity (0–15 V, increment of 1 V, 50 µs duration, interstimulus interval of 20 s). Sum action potentials were filtered (high-pass filter 3 Hz, low-pass filter 1.3 kHz) and digitized with a sampling frequency of 10 kHz. Amplitudes were determined from peak to peak.
Source: https://www.nature.com/articles/s41586-023-06090-9
Transcardial perfusion and permeabilization of mice with 4% and 4% paraformaldehyde in PBS and application to the analysis of DRGs
Mice were deeply anaesthetized and transcardially perfused with PBS (pH 7.4) followed by 4% paraformaldehyde (PFA) in PBS for 10 min. The tissues were placed in PBS for at least 1 h after dissection. The tissue was frozen on dry ice, cut in 30 m-thick free-floating sections, and stored in PBS with a dash of salt in it. Free-floating sections were permeabilized with 0.25% Triton X-100, blocked in 5%NGS for a day, and then incubated with mouse anti- NeuN at a temperature of 4 C. After washing, some sections were put into a sterilified state at 1:1,000 and Hoechst 33342. Sections were mounted with a substance. Images were acquired using Cellobserver Z1 (Zeiss) with the tile-scan module and further analysed using ImageJ.
2.5% of glutaraldehyde was perfused in PBS for TEM of tissue sections. mice were perfused with 4% and 2% PFC in the analysis of DRGs After fixing the tissues overnight, they were back to normal. The tissues were compared with osmium tetroxide, which was dehydrated. Ultrathin sections were mounted on copper grids, stained with uranyl acetate and lead citrate, and viewed by a transmission electron microscope.
Muscles freshly dissected from 2-month-old mice were fixed in 2% PFA for 15 min and subsequently washed with PBS. Fibre bundles were prepared and used for further analyses. After overnight permeabilization with 0.2% Triton X-100 in PBS, samples were blocked with 5% NGS for 1 h followed by an incubation with α-bungarotoxin-Alexa 555 (Invitrogen) 1:500 and mouse anti-NF200 overnight at 4 °C. The myofibre bundles were put to use in an experiment where they were put to use in a dilution of 1,000 for 1 h. Nuclei were stained with Hoechst 33258 (Invitrogen). Myo fibre was washed with PBS and mounted. The images were obtained using a confocal microscope and Airyscan. Images with maximum intensities are shown.
HEK293T, U2OS and HeLa cells were obtained from the American Type Culture Collection. Their identities were authenticated by STR analysis. U2OS TRex cells were provided by S. Blacklow (Brigham and Women’s Hospital and Harvard Medical School), HeLa TRex were provided by S. Taylor (Manchester University). WT, Fam134b KO and Arl6ip1 KO MEFs were isolated from embryos and immortalized using SV40 large T antigen. All cell lines were regularly tested with the Look Out Mycoplasma kit. Cells were maintained at 37 °C with 5% CO2 in DMEM medium (Gibco) supplemented with 10% FBS (Gibco) and 100 U ml–1 penicillin and streptomycin (Gibco).
Cell lines were cultured in DMEM supplemented with 10% heat-inactivated FBS and penicillin–streptomycin (Thermo Fisher Scientific) in a 5% CO2 atmosphere at 37 °C. U2OS The TRex cells provided by S. Blacklow, and the U2OS cell lines provided by the American Type Culture Collection were used to create inducible cell lines. The mCherry–eGFP–FAM134B WT and mCherry–eGFP–FAM134B 17K constructs were introduced into the vector pcDNA5/FRT/TO using GATEWAY technology and transfected with the recombinase vector pOG44 into Flp-In U2OS TRex cells. Following selection with 300 µg ml−1 hygromycin, the resistant cells were expanded. We generated U2OS cell lines expressing HA–FAM134B or HA–FAM134B 17KR under the control of a doxycycline-inducible promoter. There was a 0.25 g ml1 dose of doxycycline used for the indicated time. Lentiviruses were produced in HEK 293T cells (ATCC). In brief, HEK 293T (ATCC) cells were co-transfected with the lentiviral plasmids (1.1 µg complementary DNA (cDNA)), containing the cDNA of FAM134B WT or 17KR along with the two packaging vectors pPAX2 (2.2 µg cDNA) and pMD2.G (1 µg cDNA) using Turbofect reagent (Thermo Fisher Scientific). The medium was collected after 48 hours. The medium was placed in a container at a temperature of 80 C. After 24 h, cells were selected in fresh DMEM containing 3 µg ml−1 puromycin. Cells were maintained in the presence of antibiotics for selection and were grown to subconfluence before each experiment. The cells were treated with penicillin, chlorthalidone, cycloheximide, and 250 nM Torin 1. There are times for indicated time periods. Transient transfection was carried out using Turbofect reagent. All cell lines were regularly tested for mycoplasma contamination using the LookOut Mycoplasma PCR Detection Kit (Sigma).
Cells were collected and lysed in a buffer. 1% (w/v) NP-40, 1% (v/v) SDS, one mM. The complete protease inhibitor is a EDTA andRoche. The tissue lysates were prepared with the help of a tissue homogenizer. After sonication, homogenates were spun down at 16,900g to remove nuclei and insoluble debris. The supernatant was stored at –80 °C until further use.
The mean dark times of binding events were analysed for quantating the number of copies in the clusters. In brief, FAM134B clusters that colocalized with LC3B-II were selected manually from DNA-PAINT images and the mean dark time of binding events was determined from the plot of cumulative histograms using Picasso built-in functions. As a calibration, we used the primary and secondary antibody complexes. The inverse of dark times is the qPAINT index54 which is used for determining the number of docking strands in a cluster. We fitted the relative frequency distribution of inverse dark times with a log-normal distribution and determined the mode of distribution.
Single-molecule Localization and image reconstruction was carried out using Picasso. Single-molecule localization was achieved by integrated Gaussian maximum likelihood estimation with the following parameters: minimum net gradient = 40,000, baseline = 205, sensitivity = 4.78 and quantum efficiency = 0.95. There were gold bead markers used for the correction of drift. The point-spread function was based on the ATTO 655 single-molecule footprint with a 0.7 full width, intensity threshold and localization precision. The same origin’s signals were linked five times stronger than the nearest neighbour based analysis, with a maximum dark time of eight consecutive frames. Signals arising from the fiducial marker that passed the filtering process were removed by excluding traces from the same origin with a length exceeding 20 consecutive frames. For exchange 3D DNA-PAINT experiments, a 3D calibration curve was recorded using z-stacks (step size = 50 nm) of surface-immobilized 0.1-µm TetraSpeck Microspheres (Thermo Fisher Scientific). The section was limited to 800–1,000 nm depending on the fit. In Picasso, 3D images fromexchange DNA-PAINT experiments were aligned and visualized. TheSupplementary Video 7 is a 3D movie.
Exchange DNA- PAINT data were captured using the N-STORM super-resolution microscopy system, a 100 oil immerse objective, and an optical coherence pollutant detector from Andor Technology. The ATTO-655 conjugated oligonucleotides were excited with a collimated laser beam that was highly inclined and laminated. We acquired 20,000 frames in active frame transfer mode at 10hertz, with an gain of 200 and a pre-amp gain of one. A lens with a 10-m focal length was inserted into the emission light path to aid in 3D exchange. NIS Elements (Nikon), LCControl (Agilent) and Micro-Manager (v1.4.22)50 were used for optical setup control and data acquisition. FAM134B, LC3B-II and REEP5 were imaged sequentially following the microfluidic-assisted exchange of R2 and R1 imager strands.
The donkey anti-rabbit and goat anti-mouse were labelled with short strands of the anti-R2 strand. A 20-fold molar excess of the DBCO-sulfo-NHS ester was used to amplify the secondary antibodies. After removing excess reagent, azide-functionalized DNA-docking strands were added at a tenfold molar excess and put into a petri dish. Unbound DNA was removed from the samples concomitant with storage buffer exchange (PBS) using Amicon centrifugal filters (100 kDa cut-off). Antibody–DNA complexes were concentrated to 5 mg ml−1 and stored at 4 °C.
Molecular modeling of discontinuous DMPC and DHPC bicelles at 300 K. Simulations of bilayer-to-vesicle transitions
Discontinuous bicelle systems containing saturated DMPC (14:0 PC) and DHPC (7:0 PC) lipids were assembled as previously described4,5. The bicelle and solvated RHD molecule were obtained from simulations of the POPC bilayers. One hundred replicates for each system were simulated with different initial velocities at 300 K to obtain statistics on the transition times to vesicles. Shape transformations from flat bicelles (H = 0 nm−1) to curved vesicles (H = 0.15 nm−1) were monitored by measuring the signed membrane curvature (H(t)). The lev coordinates were fitted to the spherical surfaces using the least squares Optimization method. Positive and Negative values are reported forature away from and towards the upper/cytoplasmic leaflet. The statistics of waiting times (t) for the formation of vesicles (bilayer curvature, |H| > 0.15 nm−1) for the three systems were determined from individual replicates. The bilayer-to-vesicle transitions were modelled with a lag time of less than 15 minutes and a Poisson process. The time t′ = 1/k′ describes the Poisson process with rate k′. The constant lag time τ captures the time required for vesicle closure from the curved bilayer disc. The waiting times are defined by the distribution of them. We determined the rate of vesicle formation (k = 1/(t′ + τ)) for different systems, from fitting the cumulative distribution function for the probability density, P(t – τ) = ke−k(t – τ) corresponding to p(t), to the observed waiting time distributions estimated from replicates. For the purpose of calculating the acceleration factors we used the maximum likelihood estimates of the vesiculation rate for RHD bicelles. Furthermore, to show the effect of temperature on the energy barrier to form closed vesicles from flat bicelles, we also simulated 20 replicates of each system at 280 K and estimated the number of successful vesicle closure events within the simulation timescale.
The previously built molecular model of FAM134B RHD4 was extended to include an additional ten residues at the C terminus of the RHD (residues 261–270). Isopeptide bonds between the lysine (K160 and K264) and the terminal glycine of Ub (G76) were modelled by modifying the side-chain lysine bead (SC2/+1) into the neutral backbone bead (BB/0) and restraining its distance to the terminal bead of Ub to 0.35 nm with a force constant k = 1,250 kJ mol−1. Two mono-ubiquitinated and one bi-mono-ubiquitinated RHD structures were modelled (K160–Ub, K264–Ub and (K160 + K264)-Ub, respectively).
The transformed Escherichia coli cells were then put to use for expression and purification. In brief, the cells were grown with antibiotics overnight and used to inoculate the main culture. When the OD600 went to 0.7, 0.25 mM. isopropyl β-d-1-thiogalactopyranoside (IPTG) was used to induce protein expression for 16 h at 18 °C, shaking at 180 rpm. The cells were taken and suspended in PBS, followed by sonication and centrifugation for 30,000g. Ultracentrifugation was used to fractionated the supernatant. RHD fusion proteins were recovered by dissolving the pellets in PBS containing 0.05% dodecyl β-d-maltoside (DDM) and loading onto glutathione-Sepharose TM4 Fast Flow columns (GE Healthcare). The columns were washed with PBS containing 0.05% DDM and the proteins were eluted in PBS containing 15 mM reduced glutathione and 0.025% DDM. After sonication, theGST and theUb were loaded from the centrifugation step and then put into theGST column. The eluted fractions were concentrated and exchanged with storage buffer (50 mM HEPES pH 7.5, 150 mM NaCl and 0.0075% DDM).
Source: https://www.nature.com/articles/s41586-023-06089-2
On the Use of Human AMFR-Targeting SyRNAs for Transfection Prevention and Long-Term Detection at Dharmacon
There are human AMFR-targeting siRNAs. AGG AUC GAU UUG AAU A-3′; and siRNA #2, 5′-GUA AAU ACC GCU UGC UGU G-3′) were purchased from Dharmacon. A non-targeting siRNA was used as a control (Qiagen). The use of an siRNA was carried out usingThermo Fisher Scientific. Assays were carried out 72 h post-transfection.