The cPCDH expression regulates the organization of the neocortex
snRNA-Seq: Sequencing of 100 RNA fragments from a single hippocampal hemisphere with Cre-mCherry or Cre-GFP infection
The snRNA-seq was performed using a 10x Genomics Chromium Single Cell kit. Each reaction lane was loaded with up to 10,000 nuclei from one hippocampal hemisphere (infected with either Cre-mCherry or ΔCre-GFP) from one mouse. The manufacturer’s protocol was followed for subsequent steps for amplification and library preparation. Samples were sequenced using an Illumina NextSeq 500 with 28 bp (R1), 56 bp (R2) and 8 bp (index) reads.
The Harvard University Institutional Animal Care and Use Committee approved the use of animals. The following mouse lines were used: wild-type C57/BL6 (Jackson Laboratory, 000664); Npas4fl/fl14; Npas4-knockout14; Tip60fl/fl 49; Npas4–Flag-HA (this manuscript); Arnt2–Flag-HA (this manuscript), Tip60-H3F50; Mre11fl/fl51,52; B6;129-Gt(ROSA)26Sor
To make sure the product was a single one, the products had to be verified on DNA gels. In vitro transcription (IVT) was performed using 400 ng of purified RNA template for 16 h at 37 °C using a Megascript Short Transcript IVT kit (Thermo Fisher, AM1354). RNA clean-up was performed using a MEGAclear Transcription Clean-Up kit (Ambion, AM190). The quality was evaluated by running 200 ng of purified product on 10% TBE gels. The template was ordered as a single-stranded Ultramer from IDT with an equal number of Cortisol on either side of the cut site. CRISPR guide RNA, Cas9 RNA and Ultramer donor template were injected into zygotes of mixed background DBA/C57BL6 mice according to the guidelines and procedures of the Harvard Genome Modification facility. DNA from founders and F1 progeny were screened using Sanger sequencing to ensure proper insertion of the tag and that no mutations occurred in the flanking DNA regions. Upon request, Npas4 andFlagHA were backcrossed to C57BL6/J mice.
In order to conduct the luciferase assays, 400,000 mouse hippocampal neuron were plated onto 24 well culture dishes and transfected with plasmids using the manufacturer’s protocol. At 5 DIV, neurons were transfected with 1 µg of total DNA consisting of 450 ng of firefly luciferase reporter DNA in pGL4.11, 50 ng pGL4.74 renilla luciferase reporter DNA (Promega) and 500 ng of NPAS4 overexpression construct. The 1 g of DNA was treated with Lipofectamine. After 2 h, the transfection medium was replaced by a conditioned neuronal medium. There was a total of 1 M TTX and 100 M AP5 added during the night. At 7 DIV, neurons were collected. In brief, the neurons were washed 2 with PBS and lysed through the addition of 500 l of passive lysis buffer. Next, 20 μl of each lysate was added to one well of a Costar white polystyrene 96-well assay plate (Corning). The lysates were then added to by the linc acid Assay reagent II (LARII) and the Stop &glo reagent was used to make the luciferase/renilla measurement.
The experiments in which the seizures were caused, KA can be found in theperitoneal area. After being injected, mice were euthanized after 6 to 10 h because of previous work that shows that immediate- and late-response genes are activated after 2 and 6 h. The time point indicates the time when a mouse was injected with the same drug as the tissue it was trying to kill. For electrophysiological experiments in CA1 pyramidal neurons, mice were killed 18–24 h after injection with low levels of KA to allow sufficient time for the expression of NPAS4, its associated activity-dependent target genes and the execution of potential synaptic regulation. After 3 h of killing mice, sufficient time was given for NPAS4 to be created and for the assembly of its associatedprotein complex.
Dissected hippocampal tissue was examined under a fluorescent scope to detect GFP (ΔCre) or mCherry (Cre). Tissue that was uninfected, or in rare cases showed infection of both fluorophores in a single hemisphere, was discarded. Hippocampi were put in a buffer containing 0.25M sucrose, 25mM KCl, 5 mM MgCl2 and 20 mM Tricine-KOH. IGEPAL CA- 630 was added before being dounced and thenFILTERED through a 40- m strainer. Both mCherry and GFP were sorted on the SONY SH800 after DRAQ5 nuclear dye was added. Negative gates were determined using uninfected tissue. Nuclei were collected in 1 ml of CUT&RUN wash buffer containing 2 mM EDTA. FACS analyses in gating figures shown throughout the manuscript were performed using FlowJo (10.0.8rl).
CA1 pyramidal neurons were visualized with infrared differential interference contrast microscopy to perform whole-cell voltage-clamp recordings. Neighbouring uninfected and Cre-mCherry-infected neurons were identified using epifluorescence driven by a light-emitting diode. Neurons were held for all the experiments at 70 mV. Recording pipettes made from borosilicate glass (open resistance between 2 and 4 MΩ) were filled with an internal solution containing (in mM): 147 CsCl, 5 Na2-phosphocreatine, 10 HEPES, 2 MgATP, 0.3 Na2GTP, 2 EGTA and 5 QX-314 (Sigma-Aldrich). Internal solution was prepared in a single batch, with osmolarity adjusted to 290 mOsm with double-distilled water and pH adjusted to 7.3 with CsOH, and was stored at −20 °C. To record IPSCs, inhibitory currents were isolated through bath applications of 10 M and 10 M disodium salt. Extracellular stimulation of perisomatic inhibitory axons was achieved using a concentric bipolar electrode (FHC) placed within the centre of the stratum pyramidale and within 100–200 μm laterally of the pair of voltage-clamped cells. The stimulus strength used was the minimum stimulation required to generate a reliable IPSC in both neurons.
Recordings were made using a Multiclamp 700B amplifier (Axon Instruments), filtered at 3 kHz and sampled at 10 kHz. MatLab’s custom software was used for the data analysis. Experiments were discarded if the holding current was less than −600 pA or if the series resistance was greater than 25 MΩ. Resistance was within 25% for each pair. Recordings were performed at room temperature (19–21 °C). The amplitude of IPSCs was calculated by averaging the amplitude 0.5 ms before to 2 ms after the peak of the current. The unsigned magnitude of synaptic currents are shown for clarity.
Cryoprotection of Brain Tissue Using Transcardial Absorption and Injection of Ketamine and Xylazine
Males and females were used in the same way. Mice were anaesthetized by intraperitoneal injection of 10 mg ml–1 ketamine and 1 mg ml–1 xylazine. After anesthesia mice were transcardially perfuse with at least 10 liters of PBS and 20 liters of PFC. The brains were removed and put in 4% PFC for 24 h, followed by three washes in cold PBS. Tissue equilibrated and sank after it was immersed in 4 C of 30% sucrose for cryoprotection. The brains were stored in a frozen section medium for 30 days before being sectioned (30 m thick) on a cryostat. The parts were stored in PBS at 4 C for a while. Section were permeated and blocked for 1 h in the Blocking solution of 5% normal goat serum and 1% portobello X- 100 in PBS. The staining was performed overnight at 4 C with gentle shaking. Sections were then washed three times for 5 min each in PBS containing 0.1% Triton X-100. A rat, rabbit, and Life Technologies secondary antibodies were conjugated in a dye to produce the secondary antibody that was used for secondary staining. Sections were washed three times and mounted on a slide-scanning microscope with the 0.1% Triton X-100 and then imaged. Antibodies were diluted in blocking solution as follows: rat anti-HA (Sigma-Aldrich, ROAHAHA; 1:250); rabbit anti-NPAS4 (in house; 1:1,000)14; rabbit anti-ARNT2 (in house; 1:1,000)28; rabbit anti-KAT5 (TIP60) (Proteintech, 10827-1-AP; 1:250); rabbit anti-cleaved caspase-3 (Cell Signaling Technology, 9664S; 1:1,000).
For ChIP–seq, replicates consisted of pools of four to five mice performed on independent days. For ChIP–seq of NPAS4 from hippocampal tissue, hippocampi from 15 mice were dounced in 5 ml of 1× PBS containing protease inhibitor cocktail (Roche, 11836153001). Formaldehyde was added to tissue at room temperature with the addition of 0.125 M glycine for 5 min. For ChIP of γH2AX from Camk2a-expressing CA1 pyramidal neurons, nuclei were isolated from Camk2acre;Sun1fl/+ mice using the INTACT method53 (see ATAC-seq library preparation section for details of nuclear isolation). Following isolation, nuclei with attached beads were crosslinked with 1% formaldehyde in 1 ml of 1× PBS for 10 min at room temperature. Adding 0.25 M glycine for five minutes at room temperature quenched crosslinking. Before moving forward, crosslinked nuclei were freeze and stored at 80 C.
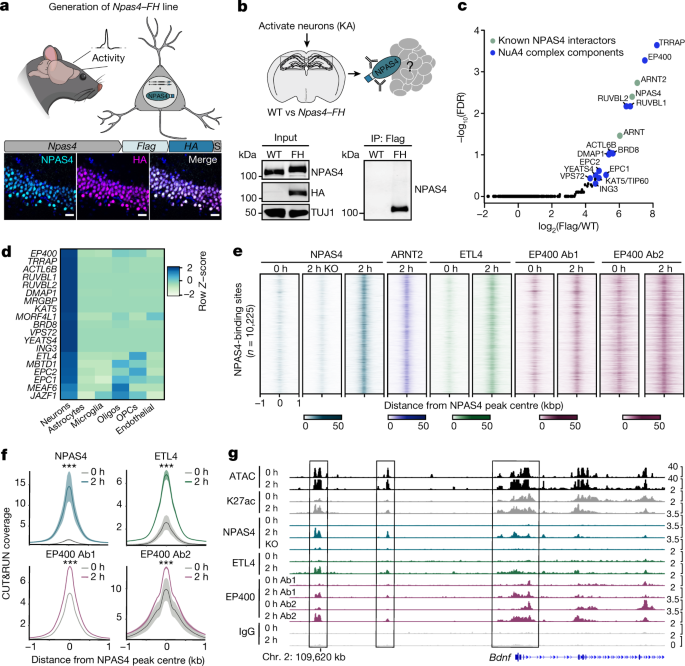
The sequence for the flag-HA-tagged FOS, JUN, EGR1, and NPAS4 was expressed in Hek293T cells. HEK293T cells were obtained from Thermo Fisher Scientific (50188404FP). HEK293T cells were not authenticated or tested for mycoplasma. With the exception of nuclear GFP-FH control samples, the plasmids expressed GFP–IRES-NPAS4 to visualize transfection efficiency. Flag-HA tags were appended to the amino terminus of NPAS4 to minimize differences in tag accessibility across NPAS4 variants truncated at the C-terminal end. Cells were collected by gently scraping into ice-cold PBS containing protease inhibitor cocktail (Roche) and were pelleted by gentle centrifugation (2,000g). Nuclei were isolated, and Flag immunoprecipitation was performed as described above. A second immunoprecipitation step using HA was performed by collecting the Flag peptide eluate, increasing the volume to 1 ml in NE1 buffer, adding 50 μl anti-HA resin (Santa Cruz Biotechnology, SC-7392 AC) and gently rotating at 4 °C for 1.5 h. Samples were washed 4× in NE1 buffer containing 250 mM NaCl for 5 min with rotation at 4 °C. Proteins were eluted by incubating resin with HA peptide (Thermo Fisher, 26184) diluted in NE1 buffer for 30 min at room temperature. Trichloroacetic acid was used for mass spectrometry. There were three days in which to perform the replicates with independent transfected cultures and immunoprecipitation assays.
Samples were processed according to standard procedures of the Taplin Mass Spectrometry Facility (Harvard University). Rehydrated proteins were incubated with 50 mM ammonium bicarbonate solution containing 12.5 ng µl–1 modified sequencing-grade trypsin (Promega) at 4 °C for 45 min. The samples were placed at 37 C in the ammonium bicarbonate solution after they were removed from the trypsin solution. Peptides were recovered through the removal of ammonium bicarbonate solution and were subsequently washed in 50% acetonitrile and 1% formic acid before dehydration.
Source: https://www.nature.com/articles/s41586-023-05711-7
Infection of Lentiviruses into Primary Neurons for ShRNA-Mediated Depletion of NPAS4 by Using a Quasi-Plasma
The following guide sequence were cloned into LentiCRIS PRv2GFP using the listed primer sequences.
To create lentiviral for shRNA-mediated depletion of NPAS4 in primary neuronal cultures or to express LentiCRISPR constructs, 10 g was transfected into HEK293T cells along with third-generation packaging plasmid. Two g of each of LentiCRISPR Fos gRNA, Scg2 gRNA, LentiCRISPR Bdnf gRNA, and LentiCRISPR Nptx2 gRNA was transfected for the pool. Following transfection, transfected medium was exchanged for fresh medium and supernatant containing viruses was collected 48 hours later. For shRNA constructs, supernatant containing virus particles from 10–15 plates of transfected HEK293T cells were pooled, and virus particles were isolated by high-speed centrifugation (25,000 r.p.m. for 90 min). In 100– 150 l of 1 PBS, the pelleted virus was resuspended overnight at 4 C. Supernatant containing virus particles from 10 plates of HEK293T cells were collected for LentiCRISPR constructs. Particles were precipitated by the addition of 1 volume of 4× PEG solution (40% PEG-8000, 1.2 M NaCl in 1× PBS pH 7.4) to 3 volumes of viral supernatant and stored for 1 h at 4 °C. Particles were precipitated by centrifugation at 1,500g for 45 min after the egg and viral mixture had been eaten. Pelleted virus was resuspended overnight at 4 °C in 500 µl of 1× PBS. In order to achieve 85% infection, each lentiviruses needed to have a minimum amount of virus of 1 l per 1.5 million cells for shRNA constructs and 60 l per 1.5 million cells for individual viruses. Neurons were infected on 3 DIV with shRNA viruses and collected at 7 DIV for RNA-seq analysis. The sBLISS-seq analysis was performed after the cells were collected at 7 DIV.
cloning and biology techniques generated the AAV backbones. AAV2/9 was prepared at the Boston Children’s Hospital Viral Core.
Source: https://www.nature.com/articles/s41586-023-05711-7
NPAS4 luciferase induction assays using quantitative PCR with high-affinity sites selected from end-repaired and tubular DNA
Luciferase induction was regulated by positioning NPAS4 target enhancers upstream of the luciferase gene. Values for the sequence to test in the assays are based on the strength of NPAS4 binding in the cells. High-affinity NPAS4 sites include regions selected from the top 100 high-confidence NPAS4-binding peaks. Regions were PCR-amplified from mouse genomic DNA and subcloned into the pGL4.11 vector using standard Gibson assembly. See below for primer sequences.
RNA was extracted using a Qiagen RNeasy Micro kit (Qiagen, 74004), and equivalent amounts of RNA (100–200 ng) across all samples were converted to cDNA using a High Capacity cDNA kit (Thermo Fisher, 4368813) according to manufacturer’s instructions. cDNA was diluted by at least threefold before running standard quantitative PCR (qPCR) with reverse transcription methods using Sybr Green master mix. The machine used for the qPCR was Applied Biosystems’ QuantStudio 3. The expression for each qPCR targetGene was normalized to Gapdh or Tubb3. The primer sets were used to amplify genes.
Sequencing libraries from precipitated DNA suspend in TE buffer were generated as previously described57, with the following changes: Rapid T4 DNA ligase (Enzymatics) was used to perform adapter ligation onto end-repaired and A-tailed DNA. The Adaptor dimers were removed from the libraries with a 1.1 ratio. There were CUT&RUN libraries that were prepared using 40-bp reads.
The number of nuclei between mice that were injected withPBS and those that were injected with KA were equal. NaCl, 0.2% Tween-20, 1 mg ml−1 BSA, 10 mM sodium butyrate and 0.5 mM spermidine supplemented with protease inhibitors). The beads were washed with a 20 mM HEPES-KOH buffer. Each sample has been added to bind the nuclei. ConA-bead-bound nuclei were incubated overnight at 4 °C in wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 0.2% Tween-20, 1 mg ml−1 BSA, 0.1% Triton X-100, 2 mM There are 10 mMsodium butyrate, 0.5 mM spermidine, and a few others in this combo.
Nuclei were release from tissue by 10 min incubation in lysis buffer 1 (LB1) (100 mM HEPES-NaOH pH 7.5, 280 mM The buffer contains 10 mM and was washed with NaCl, 2 mM equid, 2 m M EGTA, 5% Triton X-100, NP-40 and 20% glycerol. Tris-HCl pH 8.0, and 200 mM It is also known as NaCl. There were 40–42 cycles with 30-s pulses in the sonication buffer and the shear was performed with a Bioruptor. Tris-Hcl pH is 8.0. NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium deoxycholate and 0.5% N-lauroylsarcosine).
To isolate hippocampal nuclei, we placed flash-frozen hippocampal tissue in 0.5 ml of buffer HB (0.25 M sucrose, 25 mM KCl, 5 mM MgCl2, 20 mM Tricine-KOH, pH 7.8, 1 mM DTT, 0.15 mM spermine and 0.5 mM spermidine) and dounced 5× with a loose pestle and 10× with a tight pestle. IGEPAL CA-630 (5%, 32 µl) was added before douncing with a tight pestle 5–8 more times and filtering through a 40-µm strainer into a 15 ml conical collection tube. Buffer HB (3.5 ml) and 5 ml working solution (50% iodixanol, 25 mM KCl, 5 mM MgCl2, 20 mM Tricine-KOH, pH 7.8, supplemented with protease inhibitors, DTT, spermine and spermidine) were added. Homogenized tissue was gently layered on top of 1 ml of 30% iodixanol on top of a layer of 1 ml of 40% iodixanol (diluted from a working solution). Samples were centrifuged at 10,000g for 18 min, and 70 µl of nuclei was collected from the 30/40% iodixanol interface. An aliquot of each sample was incubated with trypan blue, and nuclei were counted using a standard haemocytometer.
Source: https://www.nature.com/articles/s41586-023-05711-7
Amplifying Barcoded cDNA to Detect Cre-Infections in Npas4FL Mice and Tip60FL with Q5 Gluon Polymerase
To facilitate detection of viral transcripts, 2 µl of remaining UMI-barcoded cDNA was amplified in a separate set of PCRs to increase the abundance of UMI-labelled viral transcripts. The mCherry or GFP transcripts are found in the barcoded cDNA library. There was a second reaction added to reduce nonspecific products as well as a nested strategy using Q5 high-fidelity polymerase. (R2) to the amplified product (primers: GFP amplification 1: 5′-CGCCGACCACTACCAGCAGAACACC-3′; GFP amplification 2: 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTgctgganttcgtgaccgccgcc-3′; mCherry amplification 1: 5′- CACTACGACGCTGAGGTCAAGACCACC-3′; mCherry amplification 2: 5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTgcgccgagggccgccactcc-3′). In the first and second steps of the nested PCR products were isolated using a 0.6 size selection with a reagent. The final products from the nestedPCR were then put into a sample index with the sample index primer and Chromium i7 sample index plate. These sample-indexed viral transcripts were spiked into the sequencing runs used for the corresponding full cDNA libraries. Our final data set includes data from two independent Npas4FL mice and three independent Tip60FL mice. We identified 13,536 Cre-infected and 13,486 Cre-contaminated nuclei in the Npas4FL snRNA-sq dataset, as well as 8,884 Cre- infections and 8,833 Cre infections in the Tip60FL.
To identify inducible ATAC-seq and H3K27ac peaks, we conducted a differential expression analysis using DeSeq2 (v.DESeq2_1.26.0)59 on regions that had non-zero counts in at least two of the samples. It was determined by testing against a fold change threshold of 2 and/or an increased H3K27ac CUT&RUN signal if the sites were defined as inducible elements within this set. Regulatory elements are provided in Supplementary Table 4. Note that elements that did not meet the threshold cut-off of non-zero counts in at least two of the samples will not be included in the respective DeSeq2 analysis tab in this table.
To isolate nuclei from cultured neurons plated in 6-well dishes for Cas9 control experiments, neurons were washed with ice-cold PBS to remove debris and 1 ml of HB, and 32 µl of 5% IGEPAL CA-630 was added to each well. Cells were incubated for 10 min in HB with gentle rotation at 4 °C before removal by gentle scraping and transfer to Eppendorf tubes. Nuclei from cultured neurons were fixed with 2% PFA (Electron Microscopy Sciences, 15710) followed by quenching with 125 mM glycine and gently pelleted by centrifuging at 500g. The aforementioned gradient was omitted owing to a lack of debris and myelin in neuronal cultures. After the first washed with ice-cold PBS, there were 2 more washes with ice-cold PBS. Roughly 1 106 cultured nuclei were used for the study of CRISPR–Cas9.
All experiments were sequenced on an Illumina NextSeq 500 (Illumina Next-Seq Control Software v.4.0.2). Information on sequencing data is provided in Supplementary Table 2. Single-end reads (75 bp) were obtained for ATAC-seq, ChIP–seq, RNA-seq (cultured neurons) and sBLISS-seq. Paired-end reads (40 bp) were obtained for all CUT&RUN experiments, snRNA-seq experiments and hippocampal KA time course RNA-seq experiments. Single-end reads (162 bp) were obtained for amplicon libraries used in the mutation analysis.
Primers were designed using a R script called Primer3 which is derived from a website. The 12 Ns represent the random nucleotides that make up the UMI, and the following sequence was appended onto the end of the primer. The next sequence was appended to the end of the primer. Primer sequences for the amplicons targeted can be found in Supplementary Table 5. The Primers were ordered in the form of 200 pM or 4 nM Ultramers.
Source: https://www.nature.com/articles/s41586-023-05711-7
Statistical Analysis of the Lifetimes and Peptide Quantifications of Npas4 Lickout Littermates. Experiments using NPAS4 Immunoprecipitation Mass Spectrometry
The protocols for housing knockout littermates were approved by the Harvard University Standing Committee on Animal Care. Trained technicians monitored littermates for overall health. During the data collection, distressed animals or animals with poor health were euthanized. Animals that were used for tissue collection purposes before the final collection date were censored from the final lifespan analysis. The last study ended in March 2020 due to restrictions imposed by the COVID-19 shutdown. For this reason, the lifespans of Npas4 wild-type littermates do not go to completion. The Significance of survival curves was determined using a log-rank and a Gehan–Wilcoxon–Breslow test.
The statistical analysis for each experiment is detailed in the figure legends. Electrophysiological assays were performed in a blinded manner such that the condition (saline-injected versus KA-injected) was not revealed until after the analysis was complete. Statistical methods were not used to predetermine sample sizes, but replicate numbers generally adhered to guidelines of the ENCODE consortium60. Sample randomization was not performed. All statistical analysis was performed in either Prism (v.8.4.2) or R (v.3.6.1). Multiple hypothesis correction is shown in Extended Data figs. The P values were corrected using the padjust function in base R.
Mass spectrometry and peptide quantifications were performed following the standard practices of the Taplin Biological Mass Spectrometry facility, Harvard Medical School. The data was collected using a ion-trap mass spectrometer. In brief, the program Sequest (Thermo Fisher Scientific) was used to compare peptides against protein databases with the acquired fragmentation pattern. There was a difference between a 1% and 2% false discovery rate and a reversed version of the sequence in databases. Triplicate NPAS4 immunoprecipitation–mass spectrometry experiments were performed from hippocampal tissue. Duplicate experiments were performed on high molecular weight fractions using NPAS4–Flag-HA and TIP30–H3-Flag cortical lysates. The Supplementary Table 1 contains the peptides counts for all mass spectrometry experiments. The R (3.6.1) package EdgeR61 (edgeR_3.28.1;limma_3.42.2) was used to identify proteins significantly enriched in NPAS4 or TIP60 immunoprecipitate samples relative to wild-type samples that did not express Flag-tagged proteins. Proteins that were identified in at least 2 out of 3 replicates were included in the EdgeR analysis. Peptides found strictly in wild-type control samples and not in NPAS4 or TIP60 samples (that is, background associated with the M2 resin) were removed before running the EdgeR glmFit() and glmLRT() functions.
For ATAC-seq and ChIP–seq samples (that is, NPAS4 ChIP and γH2AX ChIP), quality trimming of sequencing reads was performed with trimmomatic/0.36 (ref. 62) using the following command: java -jar trimmomatic-0.33.jar SE -threads 1 -phred33 [FASTQ_FILE] ILLUMINACLIP:[ADAPTER_FILE]:2:30:10 LEADING:5 TRAILING:5 The wind is blowing at 4:20. MINLEN:35. Nextera and TruSeq were specified for the samples. Samples were subsequently aligned to the mm10 genome using the Bowtie alignment software (vbowtie2/2.2.9) with the –very-sensitive setting. Reads mapping to the mitochondrial genome were removed. Duplicate reads were marked with Picard/2.8.0 with the command java -jar $PICARD/picard-2.8.0.jar MarkDuplicates REMOVE_DUPLICATES=false. Duplicates were subsequently removed using samtools/1.3.1 samtools view -b -F 1796.
There were significant problems for NeuN+-sorted dataset for amplicon library analysis. To remove these adapters, samples were trimmed to use the java-jar trimmomatic-0.33.jar SE -threads command. 1 -phred33 [FASTQ_FILE] ILLUMINACLIP:[ADAPTER_FILE]:2:30:10 LEADING:5 TRAILING:5 SLIDINGWINDOW:4:15 MINLEN:120. The Gene Expression Omnibus submission provides only trimmed and removed reads for the FASTQ files. Full FASTQ files will be provided upon request. Mapping using BWA was performed using the default parameters outlined in the package Debarcer v.0.3.1 (https://github.com/oicr-gsi/debarcer/releases/tag/v0.3.1)38.
Previously published32 SAR-seq datasets were downloaded from the GEO database, accession number GSE167259. Three replicates of SAR-seq performed in iNeurons (postmitotic glutamatergic neurons derived from induced pluripotent stem cells) were retrieved (GSM5100400, GSM5100401 and GSM5100402). Tag directories were used to generate aggregate plots centred on variousgenomic sites of interest. For statistical tests, signals were extracted within windows around the genomic sites of interest using homer ‘annotatePeaks.pl’ with parameters -size 500. The UCSC lifted over site lists that were generated in mice for use with human data. The sites with non overlapping signals for NPAS4 were extended to 1 kb. The NPAS4 bound sites were generated using the intersect bed -v option.
The gProfiler2 in R had a custom background of expression-filtered genes from the neuronal cell types in this manuscript, and FDR 0.05. GO terms are displayed in a graphical representation. 5e and a complete list of enriched GO terms for each cell type and dataset can be found in Supplementary Table 3.
All nuclei in the two samples were considered together for clustering. The PCA function was performed using the default parameters and the 2,000 top variable genes were identified using the FindVariable Features function. A shared nearest neighbour graph was constructed using the FindNeighbors function (considering the top 30 principal components), and clustering was assigned using the FindClusters function (resolution n = 0.02). The markers used to assign cell type to the clusters were Rbfox 3, Slc17a7, and Gad2. To assign infection status to each nucleus, we set a threshold of detecting more than eight mCherry or GFP transcripts in a given nucleus, which represented an inflection point above the background rate of detection for the distribution of these transcripts per nucleus, and reflected the expected infection patterns based on the known tropism of the AAV2/9 virus used in these experiments. The Wilcoxon rank-sum test and FindMarkers function were used to find significant genes which had an adjusted P 0.01. Heatmaps were generated using custom functions written in R. Violin plots were generated using Seurat VlnPlot function with default parameters.
In general, NPAS4 induces a diverse set of pan-neuronal and cell-type-specific effector genes. There were many targets found to be common among the 2 or 3 cell types, with a small amount being specific to one cell class. The enriched NPAS4 target genes included genes with functions in cell–cell adhesion, intercellular signalling and axon guidance as well as a variety of metabotropic and ionotropic neurotransmitter receptor subunits. There are over 1,700 new targets of NPAS4 identified by this analysis, which include genes such as Nptx2, Plk2 and Bdnf.
The homer function is used to figure out Peak annotations as enhancers or other. NPAS4 peak annotations were performed on regions extending 1 kb from the peak maxima, as these regions were most enriched for the NPAS4 motif. The peak annotations were on areas from 500 to 800 bp. The union of reproducible ATAC-seq and H3k 27ac peaks, in 3 out of three replicates, is what is considered an active regulatory element. The unions of intergenic and intronic binding site annotations are called enhancers. The sequence underlying each peak was derived using the getfasta command. The sequence of equal length for NPAS4 and MRE11, 500 bp for sBLUSS-seq were tested against the motif with the help of Meme-IP.
To determine the overlap between NPAS4 and additional factors, NPAS4 summits were extended to 1 kb. A minimum of 1 bp overlap was allowed by the bedtools/2.27.1 intersect bed. To generate sites with nonoverlapping signal for NPAS4 and FOS, NPAS4 and FOS summits were both extended to 1 kb. The peaks with NPAS4 and without were generated using bedtools/2.27.1 intersect bed -v option.
Fixed line plots were generated using homer(v4.9)’s annotatePeaks.pl [PEAK_BED] mm10 -d [INPUT_TAG_DIRS] -size 2000 –ghist -hist 25 -noann -nogene. Fixed line plots were generated from tag directories containing merged bam information from all replicates. Aggregate plots were generated using homer(v4.9)’s annotatePeaks.pl function with default parameter -hist 25, unless otherwise noted in the legend. Signal intensities were plotted using custom R scripts R (3.6.1). The average signal is shown in aggregate plots. There is a plot. The signal from all replicates was copied across the window specified in the figure legends for the statistical analysis of aggregate plots. [PEAK_BED] mm10 -d [INPUT_TAG_DIRS] with homer’s default read depth normalization to 10 million reads. Signals were averaged across all replicates, and Wilcoxon rank-sum tests were used to compare average signals between different conditions across the specified windows.
For processing the of Cre and ΔCre datasets, DeSeq2 normalization of counts was performed within each genotype (that is, raw counts across Cre versus ΔCre datasets in Npas4fl/fl were independently normalized from Cre versus ΔCre in wild-type datasets). The design we chose was because the experiments were performed at different times and there was no comparison between Cre and Cre. To account for variability in sBLISS-seq datasets generated from very low cell numbers in the Cre versus ΔCre datasets, replicates consisting of independently infected animals were not averaged on a per peak basis but rather all replicate information was retained in plotting signals (Fig. 5b and Extended Data Fig. 12a–d).
The lowest amount of reads for NPAS4 Cre and Cre and wild-type Cre were compared to the lowest number of reads for Cre. We then quantified the total number of UMIs in each sample, which is plotted in Fig. 5c and Extended Data Fig. 12e. We did not down sample inputs for DESeq2 before running it, so that we could use signals across all of the figures. The DESeq2 is used to account for the sequencing depth differences.
Using scripts in R, we calculated the mutation frequency for a given amplicon by totalling the sum of all base changes from the reference and dividing by the total number of bases assessed in the amplicon (that is, the sum of consensus depth ten families across all bases in our table). The primer anneals at the 22 bases of the sequence were omitted from analysis to make it easier to make mistakes in primer production. This calculation gives a single rate for amplicon in a sample. The total mutation rate includes both insertions and deletions and single nucleotide changes. To calculate the frequency of select point mutations, we counted the total number of select changes (C>A/(G>T)) divided by the total number of the given base included in the amplicon. The Complementary Base Changes were collapsed into a single category because it is not possible to know which strand amutation occurred. Insertion and deletion frequency was also calculated as a separate category. We calculated a per amplicon normalized mutation rate which we divided by the median number of young mice to find out the total mutations rate for each animal. For ageing gradient samples, wild-type mice aged 3 months old were considered young, 12 months old were considered middle aged and 23–27 months old were considered old. Extreme outlier points of both normalized mutation frequency and non-normalized frequency were removed across all samples using a ROUT’s test at 0.1% confidence (Fig. 5e and Extended Data Fig. 13g). Outlier removal and statistical tests on mutational samples were performed in Prism (v.8.4.2).