The tissues CD14+CD8+ T cells reprogrammed by myeloid cells
Differential cross sections and transcriptome analysis of CD14 on peripheral CD8+ T cells after PBMC and pHSC co-culture
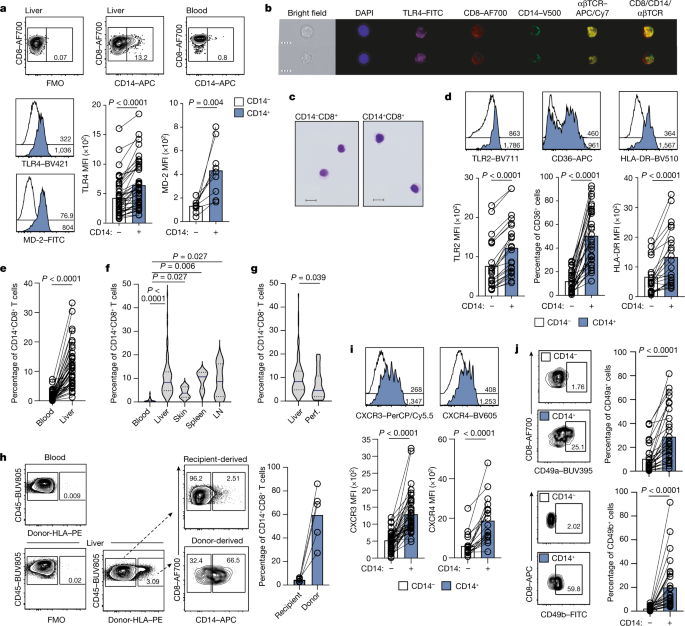
a Representative plots: %CD14 on peripheral CD8+ T cells after PBMC and pHSC co-culture (representative of four PBMC donors with 3 pHSC donors). The data was collected using pHSC Donors and PBMC donors used with pHSC. C. Summary data: MFI of TLR4 (n = 4) and TLR2 (n = 7) of pHSC-induced CD14−CD8+ T cells (black outline) or CD14+CD8+ T cells (blue filled). The peripheral CD4+T cells were cultured in 3D de-cellularised human liver ‘extracellular matrix scaffolds’. e Frequency of peripheral CD14+CD8+ T cells after co-culture of purified CD8+ T cells with a varied ratio of autologous MNP (4d; n = 11). Ex mesenchymal stem cells are isolated using a flow gating strategy that excludes debris and MNP by forward and side scatter. There is 4b and extended data. 2g–i, 3g and 4b). The results of an assessment of CD14 in FACS sorted Lin-HLA-DR+CD3-CD14+ MNP and CD14+CD8+ T cells. h Percentage expression and relative levels of T-cell genes by scRNAseq transcriptome analysis of FACS-sorted intrahepatic CD14−CD8+ T cells, CD14+CD8+ T cells or Lin−HLA-DR+CD3−CD14+ MNP (n = 2 liver samples). CD14 transcript expression and UMAP depict the transcriptomic profile of FACS-sorted withinhepatic CD14+CD8+ T cells. j Frequency of peripheral CD14+CD8+ T cells after co-culture of purified CD8+ T cells ± MNP supplemented with nocodozole (4d; n = 10). There is a CD14 MFI on CD14+CD8+ T cells after co-culture. Percentage CD14 expression on CD8+ T cells post co-cultureMDM or MNP isolated from the same donor is shown in the figure. There are representative images of purification of CD8+ T cells co-cultured with MDM and a comparison of the MFI of CD14 in the population post culture with media alone or autologous MDM. The left panel has something. The intensity of MDM-derived exosomes from donors and percentage of CD14 after co-culture is shown on the right panel. Representative plots show the function of MD2 on peripheral T-cells. CD14 and the streptavidin complex have been exposed to MNP. p Frequency of CD 14+CD8+ T cells is determined by the percentage expression in two separate regions. q Representative IMC image of macroscopically healthy liver tissue sections (2 regions of interest captured from n = 3 sections) as shown in Fig. 2n with the demarcation of three liver zones defined by intensity of CYP1A2 staining (scale bar: 200 µm; magnification: 50 µm); enrichment scores of the zonal localization of intrahepatic CD14−CD8+ T cells and CD14+CD8+ T cells. Mean and error bars can be found in the dotted lines, which represent a study participant, stained and analysed. p-values determined using either a Kruskal-Wallis test (ANOVA) with Dunn’s multiple comparisons test b (compared to media alone), j,k (compared to MNP + T-cells); or two-tailed Wilcoxon t Test c–d,l,m.
The study was approved by the National Institute of Allergy and Infectious Disease, Mount Sinai Health System, and Boston Children’s Hospital. Patients were approached in person at the hospital or by email obtained through the NIH’s DS-Connect national registry (https://dsconnect.nih.gov). The data was deidentified. The written informed consent was given for everyone in the study in compliance with the protocol of the review board. Patients with no sign of disease were collected from an outpatient routine visit where the samples were drawn. Demographics and characteristics of individuals with DS are outlined in Extended Data Tables 1 and 2. Four patients had severe disease and one had mild disease, they were all included in the data and resolved at time of sampling. Half of the patients had active disease, and three of them were in a state of treatment-free remission. From each donor, blood was drawn into a cell preparation tube with sodium heparin (BD Vacutainer). Plasma was isolated from Ficoll separation and stored at −80 °C until use. Whole blood was either directly fixed using Proteomic Stabilizer PROT1 (SmartTube) and frozen at −80 °C, or fresh whole-blood samples were stained by adding 270 µl blood directly to a MDIPA tube containing the lyophilized antibody panel, mixing and incubating for 30 min at room temperature, followed by stabilization with PROT1 and freezing.
A clinical questionnaire was filled out by patients’ legal guardian or referring physician. Each time an infectious episode occurred one point was given. There were cases in which the episode was recurrent or reported more than three times, and the score was capped at three points. 4 points are the total immune clinical score for a patient, and they would be affected by ear infections and hypothic condition.
First-principles study of the origin of IgG toxicity using the Sigma Aldrich SpinTrap method. Data analysis and analysis with FlowJo
The IgG was isolated with the help of the Sigma-Aldrich SpinTrap. The cells were put in complete RPMI and incubated with 10% FBS for 20 min or a recombinant anti-IFNGR2 antibodies (2 gml1,Thermo Fisher Scientific) at 37 C. Cells were stained with Zombie Aqua Live-Dead (BioLegend) for 30 min on ice, then fixed/permeabilized in 90% ice-cold methanol and stained with anti-phospho-STAT1-PE (1:25, BD) for 1 h on ice. Data were analysed with FlowJo after flow cytometry was acquired.
The samples were washed with PBS and then with the fluidigm. The final solution in CAS buffer consisted of 1 million cells per ml and a 1/20 dilution of EQ beads (Fluidigm). Samples were obtained at a rate of 300 events per second with a modified wide-bore injectors on the Fluidigm.
The files of acquired events were normalized with the help of theFluidigm acquisition software, and then deconvoluted to make them easier to analyse. Cell events were identified as Ir191/193-positive and Ce140-negative events. Doublets were excluded from being included on the basis of Mahalanobis distance and barcode separation. Downstream data analysis was done using the gating scheme outlined in supplementary fig. 1. The analyses were limited to adults with DS who were at least 18 years old.
Overnight, 96-well Costar plates were coated at 4 °C with 100 μl per well of a 2 μg ml−1 solution of goat anti-human IgG, F(ab′)2-fragment-specific (Jackson ImmunoResearch) or 1:100 dilution of anti-human 9G4 The IgG is suspended in 1 PBS. After the coating solution was removed the wells were washed three times with a 100 l washing buffer. Next, 200 μl of blocking buffer (PBS with 1% with bovine plasma albumin (endotoxin-free)) was added to each well at room temperature and incubated at room temperature for 1 h. The supernatants in the B cell stimulation experiments were not always very strong.
For cytokine-blocking experiments, sorted naive B cells were cultured as described above in the presence of the following blocking antibodies: anti-IFNGR2 (2 μg ml−1, Thermo Fisher Scientific, PA5-47938), anti-IFNAR2 (2.5 μg ml−1 PBL Assay Science), anti-IFNα (0.2 μg ml−1, PBL 31110–1) and IFNβ (0.2 μg ml−1, PBL 31401-1), tocilizumab (50 μg ml−1, Selleckchem), adalimumab (2 μg ml−1, Selleckchem) or a combination. The cells were stained for flow cytometry and collected.
For T cell–B cell co-cultures from HC donors and donors with DS (Fig. 4i,j), CD4 T cells were MACS-purified and co-incubated with sort-purified CD19+CD38−CD27−CD11c− B cells are on the same day. B and T cells were co-cultured for 3 days in complete RPMI supplemented with IL-21, IL-2 and anti-human IgM as above and collected for flow cytometry.
The data was created with the immunoSEQ Assay. The BCR IgH locus CDR3 has been rearranged somatically that it is amplified using a two step, amplification-bias-controlled multiplex PCR approach. The hypervariable complementarity-determining region 3 (CDR3) of the immune receptor genes is amplified with the first PCR with a forward and reverse amplification primer. A primer is included to amplify DJ rearrangements. The second PCR adds a proprietary barcode sequence and Illumina adapter sequences54. CDR3 libraries were sequenced on the Illumina instrument according to the manufacturer’s instructions.
Ficoll isolation from whole blood resulted in the clear up of plasm. According to the protocol of the manufacturer, the MILLIPLEXMAP Human Cytokine/Chemokine Magnetic Bead Panel were run. The samples were quantified on the MAGPIX xMAP Instrument (Luminex). For each sample, >50 beads were collected per analyte. The median fluorescence intensity of these beads was recorded and used for analysis. The test’s lower limit of detection for each cytokine was used when there was no signal. Patients with DS ages 7 months to 37 years and control individuals of the same age were included.
The data from the human Protein Atlas was used to make the HPAanalyze package. There was a subset from the full dataset (Supplementary Data 2) which was visualized using the ComplexHeatmap library.
GO enrichment analysis was conducted on over-abundant (log2[fold change] > 1) autoantibodies with limma’s goana function, and biological process GO terms with P < 0.0001 were subset and visualized using the ggplot263 (v.3.3.3) package.
Overnight, 96-well Costar plates were coated at 4 °C with 100 μl per well of a 1 μg ml−1 solution of recombinant IFNGR2, MSTN or ATP6V1G2 protein (OriGene) suspended in 1× PBS. The wells were washed three times the next day after the coating solution was removed. Tween 20; PBS-T). The 200 l of block buffer was put into each well at room temperature and put in a container for 1 h. The samples were nixed in a blocking buffer.
Plates were then washed three times before adding goat anti-human IgG F(ab)–horseradish peroxidase (HRP)-conjugated secondary antibody (diluted 1:3,000 in PBS-T). The plates were incubated for 1 h at 37 °C and washed three times. The plates were again washed three times with wash buffer. Once completely dry, each well was given a 100 l SigMAFAST OPD solution. This substrate was left on the plates for 5 min and the reaction was then stopped by the addition of 50 μl per well of 3 M hydrochloric acid. The plate reader utilized to measure the optical density was the POLARstar Omega.
A reporter is able to determine the blocking activity of both anti-IFn2 and anti-IFCu autoantibodies. HEK293T cells were transfected with a plasmid containing the firefly luciferase gene under the control of the human ISRE promoter in the pGL4.45 backbone and a plasmid constitutively expressing Renilla luciferase for normalization (pRL-SV40). Cells were transfected in the presence of the X-tremeGENE9 transfection reagent (Sigma-Aldrich, 6365779001) for 24 h. Cells in Dulbecco’s modified Eagle medium (DMEM; Thermo Fisher Scientific) supplemented with 2% fetal calf serum and 10% healthy control or patient serum/plasma (after inactivation at 56 °C for 20 min) were stimulated with IFNα2 (Miltenyi Biotec, 130-108-984) and IFNω (Merck, SRP3061) at 10 ng ml−1 or 100 pg ml−1 for 16 h at 37 °C. Each sample was tested once for each cytokine and dose. Finally, cells were lysed for 20 min at room temperature, and luciferase levels were measured with the Dual-Luciferase Reporter 1000 Assay System (Promega, E1980) according to the manufacturer’s protocol. Luminescence intensity was measured with a VICTOR-X Multilabel Plate Reader (PerkinElmer Life Sciences). The activity values of Firefly and Renilla were normalized.