Medin co-aggregates with Alzheimer’s disease
Ascending aortic tissue samples collected from patients undergoing elective aneurysm repair at Liverpool Heart and Chest Hospital
Ascending aortic tissue samples were obtained from patients undergoing elective aneurysmal repair at Liverpool Heart and Chest Hospital (Supplementary Table 1). This study was ethically approved by theLiverpool Bio-Innovation Hub, and all participants were given informed consent. LBIH Biobank has ethical approval to use samples through the NRES committee and is a research tissue bank. After collection, samples were rapidly frozen in dry ice and isopentane slurry, and immediately stored at –80 °C prior to use.
Amyloid load quantification in 6-month-old mice based on Congo red and anti-amyloid- staining in the neocor cortex
Amyloid-β (compact and diffuse, CAA) load was determined based on Congo red and anti-amyloid-β staining using the area fraction fractionator technique38. A set of each 36th set of systematically sampling brain sections throughout the neocor cortex was used to perform stereotypic analysis for the quantification of cortical amyloid- load. Stereo Investigator 6 is used for analysis. The Amyloid- load was calculated by the amount of anti amyloid- staining in the area. Since the 6-month-old female APP23 x Mfge8 mice only had 1–8 plaques per set of every 12th section, plaque number was quantified by blinded counting of the number of plaques rather than stereological analysis. Male 6-month-old APP23 Mfge8 WT or APP23 × Mfge8 C2 KO mice had not developed plaques yet and were therefore not included in this analysis. Stereological counts were normalized to the mean value of the wild-type mice for each age group.
The study was approved by the ethical committee of the Medical Faculty, University of Tbingen, Germany.
Male and female C57BL/6J and Mfge8 C2 knockout (C57BL/6J-Mfge8 Gt(KST227)Byg) mice15 (provided by C. Théry) were bred in-house. There are C2KO mice that were crossed with hemizygous APP mice. The APPtransgenic mouse lines used were APPPS1 (C57BL/6J-Tg(Thy1-APPK670N;M671L and Thy1-PS1L166P; generated on a C57BL6/J background)13, and APP23 (C57BL/6J-Tg(Thy1-APPK670N;M671L)14 and C57BL/6J-Tg(Thy1-APPDutch)17; backcrossed with C57BL/6J for more than 20 generations. For some experiments (such as comparisons between APP23 age groups and in vivo inoculations), mice from a separate line of APP23 mice were also used (C57BL/6JNpa-Tg(Thy1App)23/1Sdz); no differences in any measures of pathology were apparent in our experiments between these two lines (which are derived from the same founder line), as reported also previously51. Control of littermate were used in some places. All mice were maintained under specific pathogen-free conditions. The experiments that were performed were approved by the local authorities of Tbingen, Germany, and were compliant with the German veterinary office regulations.
Source: https://www.nature.com/articles/s41586-022-05440-3
Isolation and centrifuge treatment of isolated cerebral blood vessels from frozen mouse and human brain by supernatant dextran solution and BSA-HBSS
Cerebral blood vessels were isolated from frozen mouse and human brain following published protocols53,54,55,56, with small modifications. For mouse brain, 600 or 200 μl of 10% brain homogenate (see ‘Protein extraction’) were used to isolate vessels from brain. 200g of grey and white tissue was taken from frozen samples of the occipital cortex and freshly stirred up to be used in isolation of cerebral vessels. Homogenates were placed in a bucket of 2,000g for 10 min at 4 C. The supernatant was removed and mixed with an equal volume of 2× RIPA buffer (1× RIPA: There were 10 mM Tris with a pH 8.0, 1 mM EDTA, 1% Triton X-100, 10%sodium deoxycholate, and 140 mM. NaCl) freshly supplemented with protease and phosphatase inhibitors (Pierce). The pellet containing the fraction was resuspended in 18% dextran solution, mixed and thencentrifugationd at a temperature of 4 C. The pellet was resuspended in the buffer after being removed from the supernatant. The suspension was washed twice and returned from the inverted strainer with a 20 ml buffer of 1% BSA. The purified vessels were centrifuged for 20 min at 4,400g (mouse) or 2,000g (human) at 4 °C. The supernatant was aspirated, and the isolated vessels were resuspended in 1 ml BSA-HBSS and transferred to a 1.5 ml tube. In order to verify successful vessel isolation, a drop of isolated vesselssuspension was dried on Poly-d-lysine-precoated coverslips, and then stained with 4% PFA for 15 minutes at room temperature.
For extraction of insoluble protein aggregates in whole brain, RIPA-soluble and insoluble vessel fraction or isolated microglia (50,000 cells) from APP transgenic mice and human tissue, samples were treated with formic acid (Sigma-Aldrich, final concentration 70% v/v), sonicated for 30 s on ice and centrifuged at 25,000g for 1 h at 4 °C. Supernatants were then mixed with neutralization buffer (1 M Tris base, 0.5 M Na2HPO4, 0.05% NaN3 w/v; 1:20 ratio) for further downstream biochemical analysis.
There were two injections done in two female and two male APP23 mice. When possible, littermates were used to exclude any deposits of amyloid- from being observed (note that there are rarely hippocampal deposits in 9 months old APP23 mice. After anaesthesia with ketamine/xylazine (100 mg kg−1 to 10 mg kg−1 of body weight), hippocampal injections (anteroposterior, −2.5 mm; left/right, ±2.0 mm; dorsoventral, −1.8 mm) were delivered with a Hamilton syringe58 at a speed of 1.25 μl min−1. The syringe was kept in place for an additional 2 min and then slowly withdrawn. The surgical incision was closed, and the mice were closely monitored until regaining consciousness. The brains of the mice were sacrificed after 6 months of being in the lab and processed for histologic staining. Amyloid-β load was quantified stereologically (as described60; see also ‘Image analysis’).
For electron microscopy, mice were perfused with PBS, followed by a mixture of 4% PFA and 0.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4, Science Services) for 15 min. Serial frontal brain sections were cut with a vibratome (Leica VT1000S), washed in TBS, incubated in 0.1% NaBH4 (Sigma-Aldrich), and blocked with 5% BSA for 1 h at room temperature to reduce nonspecific staining. The primary and secondary anti-Mouse MFG-E8 were both used as primary and secondary anti-IgG, respectively. The sections were reacted with DAB solution at a room temperature after they were put in avidin–biotin–peroxidase complex for 90 min. The sections were silver-in intensified by the use of 3% hexamethylenetetramine and 5% silver nitrate. After staining, sections were washed in 0.1 M cacodylate buffer, osmicated (0.5% OsO4 in cacodylate buffer), dehydrated (70% ethanol containing 1% uranyl acetate (Serva)), and embedded in Durcupan (Sigma-Aldrich). The ultrathin sections were collected on Formvar- coated copper grids that were contrast-coated with lead citrate for 4 min and examined using an electron microscope.
After the resuspended vessels are removed, they must be Centrif for 10 min at 10,000g and 4 C. The resulting pellet was lysed in ice-cold 1× RIPA buffer by sonication (Bioruptor, 30 s on, 30 s off, 3 cycles, 4 °C) and shaking at 2,000 rpm at 4 °C for 15 min. After the last centrifugation for 10 min at 10,000g or 2,000g, the insoluble and insoluble fractions were separated and stored at 80 C. During the isolation,Siliconized tubes were used to increase the recovery of vessels.
The manufacturer’s instructions dictate that the levels of MFG-E8 in mouse and human samples were measured. 99% of mouse brain Homogenates were pre-diluted 1:5 and 1:10. Brain RIPA-homogenates were pre-Diluted 1:20 and RIPA-soluble vessel fractions 1: 100 for human samples. MFG-E8 levels were normalized to the total proteins content by thePierce. The data were collected on a FLUOstar Omega reader.
ThT fluorescence assays were carried out on a Flexstation 3 microplate reader (Molecular Devices). Experiments were carried out in sealed 96-well, black-walled, clear-bottomed microplates (Nunc). The data was recorded every 5 minutes using the bottom read mode. The assay was carried out using 15 µM medin (produced recombinantly as previously described62) or 20 µM Aβ40 (BioLegend) alone or co-incubated in 50 mM Tris, 150 mM 5 mM is NaCl. EDTA, pH 8 with 2 µM ThT at 37 °C under quiescent conditions with 5 s shaking before each reading. For immuno-electron microscopy of recombinant fibrils, peptides were aggregated by incubating medin and Aβ40 alone or together under the same conditions as used for ThT analysis at 37 °C for 7 days. Four-microlitre aliquots of fibrils were loaded onto carbon-coated copper grids and left for 2 min, the excess was removed by filter paper, and blocked in 1:10 goat serum in PBS, pH 8.2, containing 1% BSA, 500 ml per litre Tween-20, 10 mM For 15 min, 0.2 g l1 NaN3 and Na EDTA. The grids were then left for 2 hours at room temperature, and then washed and immuno-tacked using PBS+ and gold particles. After five 2-min PBS+ and five 2-min distilled water rinses, the grids were negatively stained using 4% uranyl acetate for 30 s. Samples were visualized on a Tecnai 10 electron microscope at 100 kV. The same protocol was used for the staining of medin aggregates isolated from human aortas and for the immuno-electron microscopy with slight modifications, including using the 1H4 antibody and a 1:10 gold particle.
Brightfield images were acquired using a Zeiss Axioplan 2 with the AxioVision 4.7 software (Zeiss) using a 4×/0.10 or 40×/0.75 objective, with fixed camera exposure time and lamp intensity for comparative stainings. The section of staining was obtained with an air 20/0.5 NA or an oil Immersion 40/1.3 NA or a 63/1.4 NA objective. Projections on maximum intensity were generated with software. All imaging and analysis steps were performed by a blinded observer.
Frequency of CAA (positive for amyloid-β and Congo Red) and the number of hemosiderin-positive microhaemorrhages was manually assessed throughout the region of interest (every 36th section in the cortex for CAA and every 12th section for hemosiderin; and every 12th section of the hippocampus, striatum and thalamus, according to previous descriptions19).
On 10- m-thick z-stacks, 30 cortical plaques and medin were analysed for co-localization. To quantify the percentage of co-localization, images were processed with Imaris (Bitplane, v. 9.7.2), and 3D surfaces were created for each channel based on a fixed intensity threshold. The overlap volume between two surfaces was measured using the function Overlapped Volume to Surfaces. The percentage of co-localization was calculated as the ratio of overlapped surface volume to total surface volume.
The plaques were obtained using the A647 or APP and LCOs at 40 magnification and the analysis was done using the z-stacks. qFTAA and hFTAA were excited with the 405 nm laser. The following functions were performed by a custom macro that analysed the images with semi-automatically predicted maximum intensity projections, including selection of plaques as regions of interest and splitting of fluorescence channels. For every plaque, plaque size and the area of the different stainings within the region of interest were then determined based on thresholded values. The qFTAA area and hFTAA staining were quantified for each plaque and the measure was determined from a ratio of qFTAA to hFTAA area. After calculating this ratio for each plaque, the mean value for all plaques per mouse was calculated.
Source: https://www.nature.com/articles/s41586-022-05440-3
Synthesis and characterization of the C-terminal medin fragment, CT-medin, using an Intavis Multipep solid-phase peptide synthesis robot
Met-Aβ was produced in-house as previously described63 using the human Met-Aβ1–42 expression plasmid, provided by C. Gomes. The C-terminal medin fragment (CT-medin: EVTGIITQGARNFGSVQFVASYK) was synthesized using an Intavis Multipep RSi solid phase peptide synthesis robot. Peptide preparations were stored as precipitates (−20 °C) and purity (>90%) was evaluated using RP-HPLC purification protocols. Peptide stocks were prepared by dissolving in appropriate buffer and filtering through 0.2-μm spin-down filters (Millipore, Germany).
The assays were carried out on a Fluostar Omega microplate reader. Experiments were carried out in sealed 96-well, half-area flat clear-bottomed microplates (Corning). Data were recorded every 10 min using bottom read mode, with excitation at 440 nm and emission at 490 nm. Prior to ThT assay experiments, lyophilised samples of Met-Aβ were suspended for 1 h at ambient conditions in 7 M guanidinium chloride in 50 mM For 5 minutes, the Tris were put through 15,000rpm at 4 C. The supernatant was injected in a Superdex 75 10/300 GL gel filtration column after equilibration. The concentration of the eluent was determined using a method known as a Nano Drop 2000 and used to calculate the eluted fraction. The peptide was diluted to a final concentration of 10μM just prior to plating. Similarly, CT-medin was dissolved in 50 mM Tris (pH 8) at a concentration of 10 μM. ThT co-aggregation and seeding assays (25 μM ThT) were run at 30 °C under quiescent conditions with 5 s shaking before each reading. For seeding assays, seeds of Met-Aβ and CT-medin were prepared by aggregating both peptides using the same protocol harvesting end-state aggregates from the plates in the absence of ThT. The end-state amyloid fibrils were sonicated for 15 min (30 s on, 30 s off) at 10 °C, using a Bioruptor Pico sonication device (Diagenode) and 0.5 µM were added to the monomeric peptides.
where fluorescence intensity (y) is represented as a function of time (x). ymax and y0 indicate maximum and starting fluorescence values, respectively, whereas t1/2 and k are the kinetic half-times and elongation rates of the fitted curves, respectively. t1/2 values were determined separately for each individual replicate per sample.
End state aggregate were prepared by running parallel assays in the absence of ThT. Independent peptide preparations (n = 3) were split into three equal aliquots before incubation and then run in triplicates, resulting in 27 total replicates. The suspensions of the peptide replicates were then mixed with pFTAA, which produced a fluorescent signal in blackplates with a clear bottom. Using a technique known as Prism 9, spatial acquisitions were background-subtracted.
Source: https://www.nature.com/articles/s41586-022-05440-3
Living Well: Statistical Analysis of Sex Effects on Mice and Steady State Microscopy in Mfge8 C2 KO Mice
Statistical analysis was done using a software. Data were tested for normal distribution (Shapiro–Wilk test) and statistical outliers were identified and removed (ROUT method), where necessary. If data were normally distributed, one- or two-way ANOVAs were performed, followed by Tukey’s multiple comparison test. We didn’t consider sex as an independent variable when we analyzed because we couldn’t detect overt sex effects. The test was performed if there wasn’t normally distributed data. For pairwise comparisons, non-parametric Mann–Whitney tests were used; all tests were two-tailed, with the exception of data on LCO ratios in Fig. 3b, where the hypothesis that the ratio would be shifted towards higher values (that is, more compact or fibrillar amyloid) in Mfge8 C2 KO mice was posited a priori based on immuno-electron microscopy data in Fig. 3a.
Linear regressions were performed using JMP software (version 14.2.0 or higher). The data must be the first log10-transformed to achieve a normal distribution. Data was analysed using afit model, which generated a number of parameters, as well as a residual and leverage plots, which displayed the statistical significance of the effect of x. The mean value of x and y is added to the residuals of y in order to calculate the data points in the graph.
Micrographs that show the staining pattern were chosen for their representation of the general pattern, which is usually reproduced at least twice. Similarly, all western blots were reproduced at least twice, with equivalent results.
This week, the Editor’s note is on a weekly round-up on living well, made simple. CNN’s Life But Better newsletter is designed to improve your well-being, so sign up for it.
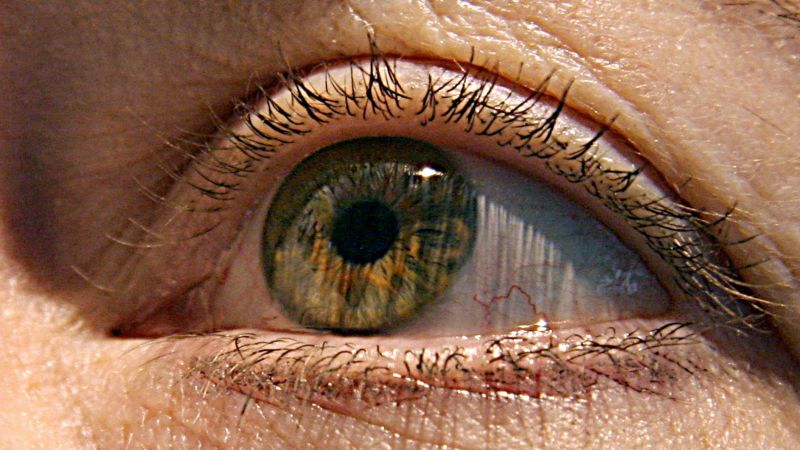
The Eye is Window into the Brain: The Molecular, Cellular, and Structural Effects of Alzheimer’s Disease in the Human Retina
“The eye is the window into the brain,” said ophthalmologist Dr. Christine Greer, director of medical education at the Institute for Neurodegenerative Diseases in Boca Raton, Florida. The back of the eye can be used to see into the nervous system.
“Our study is the first to provide in-depth analyses of the protein profiles and the molecular, cellular, and structural effects of Alzheimer’s disease in the human retina and how they correspond with changes in the brain and cognitive function,” said senior author Maya Koronyo-Hamaoui, a professor of neurosurgery and biomedical sciences at Cedars-Sinai in Los Angeles, in a statement.
“Alzheimer’s disease begins in the brain decades before the first symptoms of memory loss,” said Dr. Richard Isaacson, an Alzheimer’s preventive neurologist who is also at the Institute for Neurodegenerative Diseases.
If doctors were able to find diseases at their earliest stages, people could make healthy lifestyle choices and control their risk factors, like high blood pressure, high cholesterol and diabetes.
How early can we tell the difference between a good and bad brain? A recent study examined brain and eye tissue from people with different degrees of mental decline.
The changes in the retina correlate with changes in the brain’s temporal cortices, a hub for memory, navigation and the perception of time.
Researchers then compared samples from donors with normal cognitive function to those with mild cognitive impairment and those with later-stage Alzheimer’s disease.
The study found that cognitive issues led to the decline inglial cells. These cells are responsible for repairing and maintaining other cells, including clearing beta-amyloid from the brain and retina.
Source: https://www.cnn.com/2023/03/24/health/eye-early-alzheimers-diagnosis-wellness/index.html
The Markers of Inflammation in Plasmological Asteroseismology: A Study at the X-ray Spectrometer PHENIX
The markers of inflammation were found, and may be an equally important marker for disease progression.
Researchers found that the number of immune cells in close proximity to the plaques was higher than other cells involved in tissue death and inflammation.

