The single-molecule resolution of the function, pathology and pharmacology of the CFTR
Transduction of IMR90 and WI38 fibroblasts with the Cell Factory System at ATCC and Coriell Institute for Medical Research
IMR90 (CCL-186) and WI38 (AG06814-N) fibroblasts were purchased from ATCC and the Coriell Institute for Medical Research, respectively. The fibroblasts were grown under 8.5% CO2 and 3% O2 with 0.1 mM non-essential amino acids. Human mammary epithelial cells (HMECs; CC-2551) were purchased from Lonza and were grown under 5% CO2 and 3% O2 using the mammary epithelial cell medium complete kit (Lifeline Cell Technology, LL-0061). The number of PDs was calculated at each passage using the following equation: PD = log[number collected/seeded number]/log2. Cells have been tested to be free of mycoplasma.
shRNAs were as follows: Scramble, CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG, pLKO.1, Addgene, 1864; TRF2, ACAGAAGCAGTGGTCGAATC, pLKO.1, Open Biosystems, TRCN0000018358; shZBP1 1, CCAAGTCCTCTACCGAATGAA, pLKO.1; shZBP1 2, GCACAATCCAATCAACATGAT, pLKO.1, Sigma-Aldrich, TRCN0000123050.
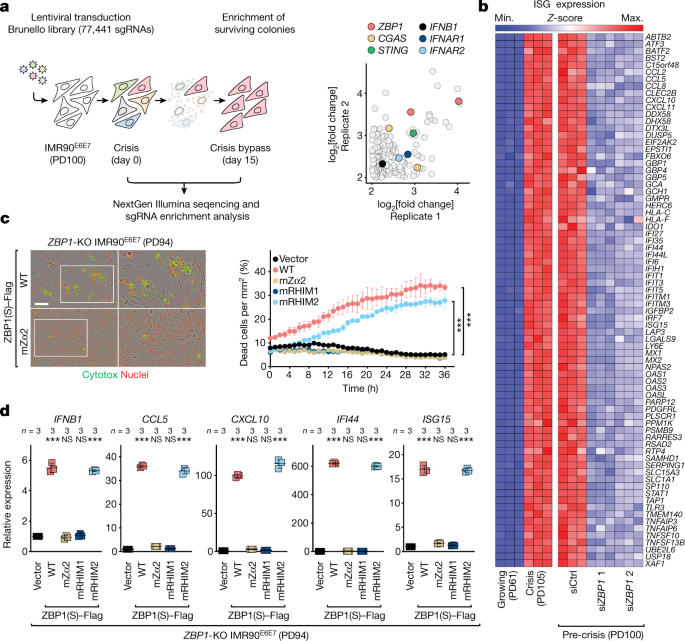
The human Brunello CRISPR knockout pooled library was obtained through Addgene. This lentiviral library has over 77 thousand gRNAs, with approximately 4 sgRNAs per Gene. For adequate representation of each sgRNA, a total of 100 million pre-crisis fibroblasts have been transduced with the lentiviral library. It was possible to perform transductions in six well plates containing 4 million cells per well, in medium containing polybrene, and centrifuging at 1,000 rcf for one hour at 33C. The next day cells were transferred to the Cell Factory System, which included puromycin-filled medium and 7 days of genome-edited cell pools. After selection, cells were pooled together and divided into two technical replicates of 30 million cells each corresponding to the library baseline control at day 0. Thirty million cells were replated into one Cell Factory System and positive selection was done for the next 15 days, which resulted in the death of a lot of cells. Two technical replicates of 30 million cells each were prepared by collecting cells at day 15, and their genomic DNA was extracted using a modified version of QIAGEN’s DNeasy Blood and Tissue Kit provided by the FLI-Seq Library Prep for CRISPR kit (Eclipse Bioinnovations). The baseline count control sample was analyzed at day 0. DNA fragments containing the sgRNA sequences were first captured from sheared gDNA, amplified by PCR using the FLI-Seq Library Prep for CRISPR kit (Eclipse Bioinnovations) and processed for next-generation sequencing. CRISPR libraries were multiplexed, normalized and pooled for sequencing. To compensate for low base diversity in libraries with high-diversity, single reads of the high-diversity libraries were done by spiking them with sequencing on the HiSeq 2500 system. Image analysis and base calling were performed using Illumina CASAVA v.1.8.2 on the HiSeq 2500 system and sequenced reads were quality-tested using FASTQC. The ratio of read count to the baseline control log was determined by the fold changes of sgRNA before and after enrichment. The Gene Set Analysis Toolkit was used for the GO enrichment analysis. ClueGO and ggplot2 R packages made it possible to see the top 20 GO terms with an FDR value of 0.05.
ZBP1(L)-, ZBP1(S)- and ZBP1(S)-deletion mutants were amplified from full-length ZBP1 gblock DNA. Fragments were cloned into BamHI-digested pLenti-CMV-MCS-(GS)5-3×Flag-Hygro using InFusion cloning.
VP16 and TRF1N were subcloned into pLPT-Empty 56. For this, VP16-TRF1ΔN and TRF1ΔN were PCR amplified from pINDUCER20-VP16–TRF1ΔN(44–439) and pINDUCER20-TRF1ΔN(44–439), respectively (both plasmids were kind gifts from P. M. Lieberman57), using either primer 5′-TGGAGAATTGGCTAGCATGGCCCCCCCGACCG-3′ or primer 5′-TGGAGAATTGGCTAGCATGCTTCTCGAGTGCCAGGTGC-3′ in combination with primer 5′-CCCCAACCCCGGATCCTCAGTCTTCGCTGTCTGAGGAAATCAG-3′. Fragments were cloned into the NheI and BamHI-digested pLPT-Empty vector using InFusion cloning.
The OMM signal ofFIS1 was fused to the C terminus of ZBCP1(S) in order to make it go to the mitochondria. The construct was cloned in three steps: first, pLenti-CMV-MCS-3×Flag-OMM-hygro was generated by amplifying the OMM of FIS1 with the primers 5′-CGATAAGGCCATCGTGGGAGGC-3′ and 5′-GAGGTTGATTGTCGACTCAGGATTTGGACTTGGACACAG-3′ from MAVS-Mito (obtained from J. Kagan through Addgene (plasmid 44556)58 and (GS)5-linker-3×Flag with the primers 5′-CGACTCTAGAGGATCCGGCTCCGGGAGCGGGTCCGGCTCTGACTATAAGGACCACGACGG-3′ and 5′-CACGATGGCCTTATCGTCATCGTCTTTGTAATC-3′ from pLenti-FNLS-P2A-GFP-PGK-Puro5. The two fragments were cloned into BamHI/SalI-digested pLenti-CMV-GFP-Hygro(656-4) (see methods of ref. cloning with InFusion Next, pLenti-CMV-MCS-(GS)5-1flag-omm-hygro was amplified by the primer 5′-CGACTCTAGG. pLenti-CMV-4flag-(GS)5- ZBP1(S)-OMM-hygro was generated by InFusion cloning using amplified fragments.
The manufacturer has instructions on how to isolated and purify totalRNA using theRNAeasy mini kit. Double digestion with the DNase I resulted in the elimination of genes. 3.5 g ofRNA was reverse-processed using the Superscript III First-Strand Synthesis System for the test, or with random hexamers for the measurement of Isg. qPCR was performed on the CFX384 Touch Real-Time PCR Detection System (BioRad). Reactions were run in triplicates with Power SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific, 4367659) in a total volume of 10 μl with standard cycling conditions. GAPDH was used as a housekeeping gene and the Comparative CT method was used to calculate relative genes. The primers are listed below.
Cells were kept at a low density of 476 cells per cm2 before being put in PBS and getting 4% paraformaldehyde. Cells were stained with 0.05% crystal violet in the water for 20 minutes.
The particles were produced in the laboratory. Production of lentivirus was performed as described previously56. In brief, HEK293T (ATCC, CRL-11268) cells were transfected with 7 µg of DNA using Lenti-X Packaging Single-Shot system (Clontech, 631276). After 48 h, the viral supernatant was collected, supplemented and used for transduction in the presence of Lenti-Blast. To produce retrovirus, Phoenix cells were transfected with 20 µg of DNA using 100 µM of chloroquine. Fresh medium was added after 5 h after transfection. The viral supernatant was collected 24 h later and used for transduction in the presence of polybrene 4 µg ml−1. Then, 48 h after infection, cells were washed and selected with 1 µg ml−1 puromycin, 600 µg ml−1 G418, or 90 µg ml−1 hygromycin. Antibiotic selection subjected fibroblasts to long-term culturing.
The Lipofectamine 3000 kit was used according to the manufacturer’s instructions. siRNA transfections were performed using the Lipofectamine RNAiMAX kit (Thermo Fisher Scientific, 13778030) according to the manufacturer’s instructions.
The cells were put into the glass covering 24 h before the experiment. Cells were fixed in 4% paraformadehyde in PBS for 10 min, washed in PBS and incubated in blocking solution of 5% BSA in PBS at room temperature. The cells were then washed in PBS and put in a room temperature tank with primary and secondary antibodies. The secondary antibodies used were AlexaFluor 488 goat anti-rabbit IgG (H+L) (Thermo Fisher Scientific, A-11034) and AlexaFluor 568 goat anti-Mouse IgG (H+L) (Thermo Fisher Scientific, A-11004). The samples were finally washed in PBS and mounted in ProLong Diamond with DAPI (Invitrogen, P36971). For staining Mito, the culture medium was added at a finalconcentration of 200 nM and then subjected to tissue culture conditions for 20 min. The cells were washed twice in prewarmed PBS, and then fixed with paraformaldehyde 4% PBS for 10 minutes. The image was performed using a microscope. Images were analysed with the help of ZEN and ImageJ.
TRF analysis was performed as previously described65. The genes were isolated and digested with MboI and AluI overnight. A total of 4 g of gDNA was separated on 0.7% agarose gel at 40 V before being transferred to a positively charged nylon membrane. After pre hybridization and after cross-linking the genes. The ssalt solution was kept for 2 h at 65 C and then used in the TelG probe. TelG probe was previously described as generating a digoxigenin labelled probe. The membranes was washed three times, with wash buffer 1 and 2, and then blocked with a blocking solution of 150 mM. NaCl, pH 7.5, 1% blocking reagent. for 30 min. Next, it was washed twice in wash buffer 3 (100M maleic acid, 150 mM) and then incubated for 30 minutes with anti-digoxigenin-AP antibodies. NaCl, pH 7.5, 0.3% (v/v) Tween-20) for 15 min each and equilibrated in AP buffer (100 mM Tris 100 mM. For two minutes, NaCl, pH 9.5. Digoxigenin-labelled telomeric DNA was detected using CDP-star ready to use (Roche, 12041677001) solution.
With minor modifications 61, the fRIP–sq was previously described. In brief, IMR90E6E7 cells were cross-linked with 0.1% formaldehyde in PBS for 10 min at room temperature and unreacted formaldehyde was then neutralized with 125 mM glycine for 5 min at room temperature. Cells were washed twice with ice-cold PBS, collected by trypsinization and the cell pellets were resuspended in RIPA buffer (50 mM Tris pH 8, 150 mM K, 0.1% SDS, 1% X-100, 5 mM. EDTA, 0.5% sodium deoxycholate, 0.5 mM DTT (added fresh), protease inhibitor cocktail (Roche, 4693159001), 100 U ml−1 RNasin Ribonuclease Inhibitor (Promega, N251B)), and incubated for 20 min at 4 °C under slow rotation. The cell lysates were centrifuged at maximum speed at 4 °C for 10 min, and the supernatants were collected and diluted in equal volumes of freshly made fRIP binding/wash buffer (150 mM KCl, 25 mM Tris pH 7.5, 5 mM EDTA, 4.6% NP-40, 0.25 mM DTT and 100 U of 1 RNasin ribonuclease Inhibitor (Promega, N251B). The lysates are precleared with Dynabeads and then incubated with 10 g under a slow rotation for 30 min. The beads were washed twice in a buffer with a fRIP binding/wash buffer and then reversed in a 3 PBS buffer. EDTA, 15 mM DTT (added fresh), 10 μl of 20 mg ml−1 proteinase K (Millipore, 70663) and 1 μl of RNasin Ribonuclease Inhibitor. The reverse-cross-linking was done at 55 C for 1 h and 42 C for another hour. The Direct-zol RNA Microprep kit is recommended by the manufacturer and can be used for the recovery of nucleic acids.
Total RNA was isolated using TRIzol (Invitrogen, 15596018) and purified using the RNAeasy Mini Kit (Qiagen, 74106) according to the manufacturer’s instructions. Genomic DNA was eliminated by double digestion with DNase I (RNase-free DNase Set, Qiagen, 79256). The quality of the isolated total RNA was assessed using the Agilent TapeStation 4200 and RNA-seq libraries were prepared with 500 ng total RNA using the TruSeq stranded mRNA sample preparation kit according to the manufacturer’s protocol (Illumina). Multiplex, normalized and pooled were used for the seq libraries. The libraries were sequenced on the HiSeq 4000 system (Illumina) as 50 bp single reads or the NextSeq 500 system (Illumina) as 75 bp single reads. Image analysis and base calling were performed using Illumina CASAVA v.1.8.2 on the HiSeq 4000 system and sequenced reads were quality-tested using FASTQC. Sequenced reads were quality-tested using FASTQC (v.0.11.8)68 and mapped to the hg19 human genome using the STAR aligner (v.2.5.3a)62 with the default parameters. Using analyzeRepeats.pl, the top expressed isoform was used as a proxy for the transcript expression of RefSeq genes. To compute within-group dispersion using replicates, differential gene expression was done on the raw gene counts with the R package. When compared to two experimental conditions, the differentially expressed genes were defined as having FDR 0.05 and a log2[fold change] > 0.585. GO enrichment analysis was performed using the WEB-based Gene Set Analysis Toolkit” (WebGestalt) and the R package clusterProfiler. The ClueGO and ggplot2 R packages were used to show the importance of the 20 GO terms with an FDR value of 0.05. Heat maps and Venn diagrams were generated using the R packages ComplexHeatmap and VennDiagram, respectively.
A million cells were put into a 10 cm plate for 24 h and then culture medium was collected andcentrifugations performed to remove debris. There are three technical replicates of 50 l each. For the preparation of standards, 0.1 mM of non-essential Amino Acids was added to the GlutaMax-DMEM. IFNβ secretion in cell culture supernatant was quantified using the IFNβ enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems Human IFNβ Quantikine ELISA Kit, DIFNB0) according to the manufacturer’s instruction. Supernatants from the treated cells were collected and incubated in IFNβ ELISA kit for 2 h at room temperature and washed three times with wash buffer. The optical density was measured with a microplate reader and data was plotted against a standard curve to determine the concentration of IFN.
R&D Systems, 8499-IF-020/CF and InvivoGen were the names of the reagents. Cell Signaling Technology, 60332 is a cell signaling technology. nigericin is found in the Cayman Chemical. Thermo Fisher Scientific, 62249.
Unpaired two-tailed Student’s t-test for vector versus ZBP1(S); siCtrl versus siIRF3 (S) and siPYCARD
Statistical analysis was performed using Prism 9. Comparisons between two groups were performed using unpaired two-tailed Student’s t-tests. Multiple comparisons were performed using one-way ANOVA followed by Tukey’s or Dunnett’s multiple-comparisons test. Results from two or three independent experiments were obtained for all representative findings. Significance in all figures is denoted as follows: P < 0.05, P < 0.01, **P < 0.001.
Extended Data Fig. 6e: vector versus ZBP1(S)–Flag (0.0082); siCtrl versus siZBP1 (<0.0001); siCtrl versus siRIPK3 (0.9942); siCtrl versus siMLK (>0.9999); siCtrl versus siRIPK1 (>0.9999); siCtrl versus siFADD (0.9425); siCtrl versus siCASP8 (>0.9999); siCtrl versus siNLRP3 (>0.9999); siCtrl versus siPYCARD (>0.9999); siCtrl versus siCASP1 (0.9790); siCtrl versus siIFNAR1 (<0.0001); siCtrl versus siIFNAR2 (<0.0001); siCtrl versus siSTAT1 (<0.0001); siCtrl versus siSTAT2 (<0.0001); siCtrl versus siIRF9 (<0.0001); siCtrl versus siATG5 (<0.0001); siCtrl versus siATG7 (<0.0001); siCtrl versus siATG12 (<0.0001).
Extended Data Figure 11c: IMR90E6E7: TOM20–ZBP1, growing versus crisis (<0.0001); MitoTracker–ZBP1, growing versus crisis (<0.0001); TFAM–ZBP1, growing versus crisis (<0.0001). WI38SV40LT: TOM20–ZBP1, growing versus crisis (<0.0001); MitoTracker–ZBP1, growing versus crisis (<0.0001); TFAM–ZBP1, growing versus crisis (<0.0001).
Figure 4a: PD35 versusPD62 and PD45 versusPD99 both had 0.0001 outcomes.
Extended Data Fig. 3d shows IMR90E6E7, after 24 h of incubateting with clitox. sgGFP versus sgZBP1 2 (<0.0001); sgGFP versus sgZBP1 3 (<0.0001); sgLUC versus sgZBP1 1 (<0.0001); sgLUC versus sgZBP1 2 (<0.0001); sgLUC versus sgZBP1 3 (<0.0001). WI38SV40LT, 24 h after incubation with Cytotox. sgGFP versus sgZBP1 1 (<0.0001); sgGFP versus sgZBP1 2 (<0.0001); sgGFP versus sgZBP1 3 (<0.0001); sgLUC versus sgZBP1 1 (<0.0001); sgLUC versus sgZBP1 2 (<0.0001); sgLUC versus sgZBP1 3 (<0.0001).
Extended Data Fig. 4e: The experiment is to see which one will give the better result; in this case, the experiment is to see which one will yield the better result; in this example, the experiment is to see which one will yield the better result. SiCtrl vs Si CGAS and siAIM2 in the second Experiment. The experiment 3 involved the relationship between siCtrl and SiCGAS.
The Data is extended by mock versus shScraamble day 3, mock versus shScraamble day 6 and mock versus shScraamble day 9.
Extended Data Figure 11e: dsRNA intensity: siCtrl versus siSUV3 (<0.0001); siCtrl versus siPNPase (<0.0001); number of ZBP1 filaments: siCtrl versus siSUV3 (0.6179). siCtrl versus siPNPase (0.3240).
There were two pools of siRNAs, the non-targeting one and the targeted one. GGAUUUCCAUUGCAAACUC; ZBP1 4 (Dharmacon, J-014650-05-0005): CAAAGUCAGCCUCAAUUAU; MB21D1 (Dharmacon, L-015607-02-0005): GAAGAAACAUGGCGGCUAU, AGGAAGCAACUACGACUAA, AGAACUAGAGUCACCCUAA, CCAAGAAGGCCUGCGCAUU; TMEM173 (Dharmacon, L-024333-02-0005): UCAUAAACUUUGGAUGCUA, CGAACUCUCUCAAUGGUAU, AGCUGGGACUGCUGUUAAA, GCAGAUGACAGCAGCUUCU; MAVS 1 (Dharmacon, J-024237-07-0005): GCAAUGUGGAUGUUGUAGA; MAVS 2 (Dharmacon, J-024237-05-0005): AAGUAUAUCUGCCGCAAUU; MAVS 3 (Dharmacon, J-024237-06-0005): CAUCCAAAGUGCCUACUAG; MAVS 4 (Dharmacon, J-024237-08-0005): CAUCCAAAUUGCCCAUCAA; PYCARD (Dharmacon, L-004378-00-0005): GGAAGGUCCUGACGGAUGA, UCACAAACGUUGAGUGGCU, GGCCUGCACUUUAUAGACC, CCACCAACCCAAGCAAGAU; MYD88 (Dharmacon, L-004769-00-0005): CGACUGAAGUUGUGUGUGU, GCUAGUGAGCUCAUCGAAA, GCAUAUGCCUGAGCGUUUC, GCACCUGUGUCUGGUCUAU; TLR7 (Dharmacon, L-004714-00-0005): CAACAACCGGCUUGAUUUA, GGAAAUUGCCCUCGUUGUU, GAAUCUAUCACAAGCAUUU, GGAAUUACUCAUAUGCUAA; TLR3 (Dharmacon, L-007745-00-0005): GAACUAAAGAUCAUCGAUU, CAGCAUCUGUCUUUAAUAA, AGACCAAUCUCUCAAAUUU, UCACGCAAUUGGAAGAUUA; TLR9 (Dharmacon, L-004066-00-0005): CAGACUGGGUGUACAACGA, GCAAUGCACUGGGCCAUAU, CGGCAACUGUUAUUACAAG, ACAAUAAGCUGGACCUCUA; TLR8 (Dharmacon, L-4715-00-0005): CAACGGAAAUCCCGGUAUA, CAGAAUAGCAGGCGUAACA, GUGCAGCAAUCGUCGACUA, CUUCCAAACUUAUCGACUA; DDX58 (Dharmacon, L-012511-00-0005): GCACAGAAGUGUAUAUUGG, CCACAACACUAGUAAACAA, CGGAUUAGCGACAAAUUUA, UCGAUGAGAUUGAGCAAGA; IFIH1 (Dharmacon, L-013041-00-0005): GAAUAACCCAUCACUAAUA, GCACGAGGAAUAAUCUUUA, UGACACAAUUCGAAUGAUA, CAAUGAGGCCCUACAAAUU; TICAM1 (Dharmacon, L-012833-00-0005): GGAGCCACAUGUCAUUUGG, CCAUAGACCACUCAGCUUU, GGACGAACACUCCCAGAUC, CCACUGGCCUCCCUGAUAC; IFNAR1 (Dharmacon, L-020209-00-0005): GCGAAAGUCUUCUUGAGAU, UGAAACCACUGACUGUAUA, GAAAAUUGGUGUCUAUAGU, GAAGAUAAGGCAAUAGUGA; IFNAR2 (Dharmacon, L-015411-00-0005): CAGAGGGAAUUTUUAAGAA, GAGUAAACCAGAAGAUUUG, CACCAGAGUUUGAGAUUGU, UCACCUAUAUCAUUGACAA; AIM2 (Dharmacon, L-011951-00-0005): GCACAGUGGUUUCUUAGAG, UCAGACGAGUUUAAUAUUG, GAAAGUUGAUAAGCAAUAC, GUUCAUAGCACCAUAAAGG; SUPV3L1 (Dharmacon, L-017841-01-0005): UGGCUAAGCUACCGAUUUA, GUAAGGAUGAUCUACGUAA, CGGUGCAGCUCAUGCGGAU, GGAAAGACUUAUCACGCAA; PNPT1 (Dharmacon, L-020112-00-0005): GACAGAAGUAGUAUUGUAA, ACAGAAAGAUUAUUGGCUA, GAAUGUAAGUUGUGAGGUA, AAUCAGAGAUACUGGUGUA; RIPK3 (Dharmacon, L-003534-00-0005): CCACAGGGUUGGUAUAAUC, AACCAGCACUCUCGUAAUG, GCUACGAUGUGGCGGUCAA, GACCGCUCGUUAACAUAUA; ATG7 (Dharmacon, L-020112-00-0005): CCAACACACUCGAGUCUUU, GAUCUAAAUCUCAAACUGA, GCCCACAGAUGGAGUAGCA, GCCAGAGGAUUCAACAUGA; TERRA 1 (Dharmacon, CTM-536949): AGGGUUAGGGUUAGGGUUAUU; TERRA 2 (Dharmacon, CTM-536950): GGGUUAGGGUUAGGGUUAGUU; MLKL (Dharmacon, L-005326-00-0005): GAGCAACGCAUGCCUGUUU, CAAACUUCCUGGUAACUCA, GAAGGAGCUCUCGCUGUUA, GGAUUUGCAUUGAUGAAAC; RIPK1 (Dharmacon, L-004445-00-0005): CCACUAGUCUGACGGAUAA, UGAAUGACGUCAACGCAAA, GCACAAAUACGAACUUCAA, GAUGAAAUCCAGUGACUUC); FADD (Dharmacon, L-003800-00-0005): CAUUUAACGUCAUAUGUGA, GGAGAAGGCUGGCUCGUCA, UGACAGAGCGUGUGCGGGA, GCAUCUACCUCCGAAGCGU); CASP8 (Dharmacon, L-003466-00-0005): GGACAAAGUUUACCAAAUG, GCCCAAACUUCACAGCAUU, GAUAAUCAACGACUAUGAA, GUCAUGCUCUAUCAGAUUU); NLRP3 (Dharmacon, L-017367-00-0005): GGAUCAAACUACUCUGUGA, UGCAAGAUCUCUCAGCAAA, GAAGUGGGGUUCAGAUAAU, GCAAGACCAAGACGUGUGA); CASP1 (Dharmacon, L-004401-00-0005): GGAAGACUCAUUGAAACAUA, GAUGGUAGAGCGCAGAUGC, CCGCAAGGUUCGAUUUUCA, GAGUGACUUUGACAAGAUG); STAT1 (Dharmacon, L-003543-00-0005): GCACGAUGGGCUCAGCUUU, CUACGAACAUGACCCUAUC, GAACCUGACUUCCAUGCGG, AGAAAGAGCUUGACAGUAA); STAT2 (Dharmacon, L-012064-00-0005): GGACUGAGUUGCCUGGUUA, GGACUGAGGAUCCAUUAUU, GAGCCCUCCCUGGCAAGUUA, GAUUUGCCCUGUGAUCUGA); IRF9 (Dharmacon, L-020858-00-0005): GCAGAGACUUGGUCAGGUA, CCACCGAAGUUCCAGGUAA, GCGUGGAGCUCUUCAGAAC, GAAAGUACCAUCAAAGCGA); ATG5 (Dharmacon, L-004374-00-0005): GGCAUUAUCCAAUUGGUUU, GCAGAACCAUACUAUUUGC, UGACAGAUUUGACCAGUUU, ACAAAGAUGUGCUUCGAGA); ATG12 (Dharmacon, L-010212-00-0005): GAACACCAAGUUUCACUGU, GCAGUAGAGCGAACACGAA, GGGAAGGACUUACGGAUGU, GGGAUGAACCACAAAGAAA).
The primers were as follows: 9p-F, GAGATTCTCCCAAGGCAAGG; 9p-R, ACATGAGGAATGTGGGTGTTAT; 13q-F, CTGCCTGCCTTTGGGATAA; 13q-R, AAACCGTTCTAACTGGTCTCTG; 15q-2-F, CAGCGAGATTCTCCCAAGCTAAG; 15q-2-R, AACCCTAACCACATGAGCAACG; Xq-F, AGCAAGCGGGTCCTGTAGTG; Xq-R, GGTGGAACTTCAGTAATCCGAAA; Xp-F, AAGAACGAAGCTTCCACAGTAT; Xp-R, GGTGGGAGCAGATTAGAGAATAAA; GAPDH-F, AGCCACATCGCTCAGACAC; GAPDH-R, GCCCAATACGACCAAATCC; GAPDH RT, GCCCAATACGACCAAATCC; TERRA RT, CCCTAACCCTAACCCTAACCCTAACCCTAA; ZBP1-F, AACATGCAGCTACAATTCCAGA; ZBP1-R, AGTCTCGGTTCACATCTTTTGC; IFNB1-F, ACGCCGCATTGACCATCTAT; IFNB1-R, GTCTCATTCCAGCCAGTGCT; ISG15-F, CGCAGATCACCCAGAAGATCG; ISG15-R, TTCGTCGCATTTGTCCACCA; IFI44-F, AGCCGTCAGGGATGTACTATAAC; IFI44-R, AGGGAATCATTTGGCTCTGTAGA; CCL5-F, CCAGCAGTCGTCTTTGTCAC; CCL5-R, CTCTGGGTTGGCACACACTT; CXCL10-F, GTGGCATTCAAGGAGTACCTC; CXCL10-R, TGATGGCCTTCGATTCTGGATT.
Growth and Labelling of a Human CF.TR Baculovirus in a Freestyle SF-900 SFM Medium with Heat-Inactivated Feto-Bovine Semen and Antimicrobial-Antimy
As described previously, it was termed CF.TR. Human CFTR with a C-terminal PreScission Protease-cleavable GFP tag was cloned into the BacMam vector. The following replacements were added to single-molecule FRET.
Recombinant baculovirus was generated using Sf9 cells (Gibco, catalogue number 11496015, lot number 1670337) cultured in sf-900 SFM medium (Gibco), supplemented with 5% (v/v) heat-inactivated fetal bovine serum and 1% (v/v) antibiotic–antimycotic (Gibco) as described previously48. suspension cells were cultured in freestyle 293 medium with 2% heat-inactivated fetal bovine semen and 1% antibiotic Sf9 and HEK293S GnTI− cells were authenticated by Gibco and ATCC, respectively and confirmed negative for mycoplasma contamination. 10% of the cells were found to have P3 baculoviruses, and the density was 2.5 106 cells. After 12 h, the culture was supplemented with 10 mM sodium butyrate, and the temperature was reduced to 30 °C. After a further 48 h, the cells were collected and flash-frozen in liquid nitrogen.
To label with conjugated maleimide, 10 M maleimide and 10.5 M maleimide were mixed together. Subsequent steps were carried out protected from light. The labelling reaction was quenched by addition of 2 mM DTT, and the labelled product was purified by gel filtration chromatography at 4 °C using a Superose 6 10/300 GL column (GE Healthcare), equilibrated with 0.06% (w/v) digitonin, 200 mM NaCl, 20 mM HEPES (pH 7.2 with NaOH), 1 mM ATP and 2 mM MgCl2. Peak fractions were concentrated to 2 μM and mixed with 5 μM biotin–tris-NTA-Ni2+ for 30 min at 4 °C. After another round of gelfiltration, 2 M, snap-frozen in liquid nitrogen, and stored at -80 C, theCFTR–Ni-NTA complex was re-purified.
The buffer for the purification process was changed from 1.25% to 0.25% and 200 mM to 20 mM. PMSF and 3 µg ml−1 DNase I. Wash and gel filtration buffers contained 0.06% (w/v) digitonin, 20 mM HEPES (pH 8.0 with KOH), 200 mM KCl, 2 mM MgCl2 and 2 mM DTT. The eluate from the GFP is concentrated, phosphorylated with NEB for 1 h, and immediately used for hydrolysis measurements.
There were Chinese hamster ovary cells maintained in a mixture of heat-inActivated Fetal bovine serum and 1% Glutamax. Chinese hamster ovary cells were authenticated by ATCC. 24 h before transfection, the cells were plated in 35-mm cell culture dishes. Cells were transfected with GFP-fused genes cloned into the BacMam expression vectors, using a vaccine. At 12 h following transfection, medium was replaced with DMEM-F12 supplemented with 2% (v/v) heat-inactivated fetal bovine serum and 1% (v/v) GlutaMAX, and the cells were then incubated for 24 h at 30 °C before recording.
The solution for the bath contained 145 mM. NaCl, 2 mM MgCl2, 5 mM KCl, 1 mM CaCl2, 5 mM glucose, 5 mM HEPES and 20 mM sucrose (pH 7.4 with NaOH). 140 mM of NMDG was contained in the pipette solution. 2 mM HePES (pH 7.4 with HCl) is used for CaCl2, 2 mM MgCl2 and 10 mM HEPES. The solution contained 150 mM. NMDG, 2 mM MgCl2, 1 mM CaCl2, 10 mM EGTA and 8 mM The tris were 7.4 with HCl. Where it was indicated, magnesium was not included. CFTR was activated by exposure to PKA (Sigma-Aldrich) and 3 mM ATP.
The rate of buffer exchange by the perfusion system was estimated by exchanging perfusion solution with 150 mM NMDG, 2 mM MgSO4, 1 mM calcium gluconate, 10 mM EGTA and 8 mM Tris (pH 7.4 with H2SO4).
Pipettes were pulled from borosilicate glass (outer diameter 1.5 mm, inner diameter 0.86 mm, Sutter) to 1.5–2.5 MΩ resistance and fire polished. Recordings were carried out using the inside-out patch configuration with local perfusion at the patch. The potential was reached at 30 mV and the currents were recorded at 25 C. Recordings were low-pass-filtered at 1 kHz and digitized at 20 kHz. All displayed recordings were further low-pass filtered at 100 Hz. Data were analysed with Clampfit, GraphPad Prism and OriginPro.
Source: https://www.nature.com/articles/s41586-023-05854-7
Reactivation of proteoliposome mixtures containing CFTR and methylated beta-cyclodextrin
A lipid mixture containing 1,2-dioleoyl-sn-glycero-3-phosphoetanolamine, 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine at a 2:1:1 (w/w/w) ratio was resuspended by sonication in buffer containing 200 mM There are 20 mM HePES with NaOH and 2 mM-M MgCl2. There was a final detergent concentration of 2% mixed with GDN and the finallipid concentration was 20 ng/L covered by argon gas. Purified CFTR was mixed with the lipid mixture at a protein-to-lipid ratio of 1:100 or 1:250 (w/w) and incubated at 4 °C for 2 h covered by argon gas. Methylated beta-cyclodextrin was added to the reaction at a 1.2× molar ratio to GDN. The equivalent amount of the drug was added after an additional 4 h. This procedure was repeated for a total of four additions. After being centrifugation for 45 min, proteoliposomes were resuspended in buffer with 200 mM. Aliquoted, snap-frozen and stored at 80 C are the contents of the NaCl, 20mM HePES and 2 mM MgCl2.
The vesicles used for the image of proteoliposome-reconstituted CFTR were made through a process called polylactic acid (PLA). The vesicles were then incubated with 1 µM biotin–tris-NTA-Ni2+. Excess biotin–tris-NTA-Ni2+ was removed by pelleting the vesicles by ultracentrifugation at 150,000g for 45 min, removing the supernatant and resuspending in buffer containing 150 mM Two mM HEPES, two mM NaCl and one mM NaOH are listed. The procedure was repeated a second time. The sicles were immobilized for five minutes in the chambers and then the segulls could be washed with buffer. The deoxygenated buffer held 150 mM. NaCl, 2 mM MgCl2, 20 mM HEPES (pH 7.2 with NaOH), 2 mM protocatechuic acid and 50 nM protocatechuate-3,4-dioxygenase.
Single-molecule imaging was carried out using a custom-built wide-field, prism-based total internal reflection fluorescence microscope. LD555 fluorophores were excited with an evanescent wave generated using a 532-nm laser (Opus, Laser Quantum). Emitted fluorescence from LD555 and LD655 was collected with a 1.27 NA 60× water-immersion objective (Nikon), spectrally separated using a T635lpxr dichroic (Chroma), and imaged onto two Fusion sCMOS cameras (Hamamatsu) with integration periods of 10 or 100 ms.
Single-molecule fluorescence data were analysed using SPARTAN analysis software in MATLAB21. FRET trajectories were calculated from the emitted donor and acceptor fluorescence intensities (ID and IA, respectively) as EFRET = IA/(IA + ID). FRET trajectories were selected for further analysis on the basis of the following criteria: single-step donor photobleaching; a signal-to-noise ratio >8; fewer than 4 donor-blinking events; and FRET efficiency above baseline for at least 50 frames. Further, single-molecule traces exhibiting FRET values above 0.8 were excluded from analysis. This subpopulation was insensitive to phosphorylation and nucleotide, and probably reflected denatured molecules. Humans manually curated traces to remove obvious photophysical artefacts for kinetic analysis. FRET trajectories were idealized using the segmental k-means algorithm52 with a model containing two non-zero-FRET states with FRET values of 0.25 ± 0.1 and 0.48 ± 0.1. Data were further analysed with GraphPad Prism and OriginPro.
The dephosphorylated wild-type CF TR was concentrated to less than one half of one liter. Concentrations of 3 mM ATP and 3 mM fluorinated Fos-choline-8 were added to the sample immediately before application onto Quantifoil R1.2/1.3 400 mesh Au grids and then vitrification using a Vitrobot Mark IV (FEI).
Source: https://www.nature.com/articles/s41586-023-05854-7
Refinement of Gatan K2 Summit Micrographs and GCTF55 with Second-Order Correction for Beam-Induced Motion
The images were collected using a Gatan K2 Summit detector and a 300-keV Titan Krios transmission electron microscope. The defocus range of the micrographs was 0.8– 2.5 m, with a physical size of 1.03. Micrographs were recorded with 10-s exposure (0.2 s per frame) with a dose rate of 8 electrons per pixel per second.
Image stacks were gain-normalized, binned by 2, and corrected for beam-induced specimen motion with MotionCor2 (ref. 54). Contrast transfer function estimation was carried out using GCTF55. Images with estimated resolutions below 4.5 Å were removed. Particles were initially picked with the Laplacian-of-Gaussian implementation in RELION56. The selected two-dimensional classes from this set were used to pick particles. The particles were cleaned by a process that used two- and three-dimensional classification. A total of 157,629 particles were included in the final refined map.

