The FG phase of nuclear pores is where HIV-1 capsids enter
Measurement of the order and disorder of nups and peptides obtained from GFP-Nup and AF488 cells in a scanning stage
FFS was performed as described by Lau et al.17. Briefly, cell-free expressed GFP-Nup proteins (approximately 50 nM; equivalent to approximately 2,500 photon count) or AF488 peptides (100 nM) were mixed with AF568-HIV-1 CA assemblies (12 μM) in 50 mM Tris, pH 8, 150 mM What is NaCl? A scanning stage operated at 1m s1 recordedescence traces for 15 s per trace in 1 ms bins. Typically, measurements were repeated 10–15 times per sample.
The order and disorder57 were determined by the per-residue relative solvent accessibility calculated from AlphaFoldDB structures. RSA-based order and disorder for all Nups were accessible through Mobi-DB58.
Source: The HIV capsid mimics karyopherin engagement of FG-nucleoporins
Importin- and IBB-mCherry in pET11a (GenScript) as His-fusion protein in e. coli Rosetta2 cells
The mCherry protein was kindly provided by J. Goyette. Importin-β and IBB-mCherry in pET11a (GenScript) were expressed as His-fusion proteins in E. coli Rosetta2 cells. The cells were grown at 37 C and had an expression of 1.5 mM. Cell growth was continued at 18 C with an optical density of 0.6. Cells were collected (4,000g, 10 min, 4 °C), and the cell pellets were resuspended in lysis buffer (purification buffer: 20 mM The Tris has a pH of 7.5 and 150 mM. NaCl, 2 mM DTT; +cOmplete EDTA-free Protease Inhibitor (Roche)) and lysed by sonication. The lysate was cleared bycentrifugation and then the supernatant was bound to Ni-beads in a purification buffer. The beads were washed with 20 column volumes of wash buffer (purification buffer + 20 mM imidazole) and eluted in 2 ml fractions with elution buffer (purification buffer + 500 mM imidazole). Protein-containing fractions were pooled, and cleavage of the His-tag was achieved with TEV (IBB-mCherry) or SUMO (importin-β) in overnight dialysis against TEV-cleavage buffer (50 mM Tris pH 7.5, 1 mM DTT) or SUMO-cleavage buffer (50 mM The tris has a pH of 7.5. 1 mM DTT is NaCl. The flow-through was collected after cleaving proteins were added to Ni-beads. Proteins were finally purified by size-exclusion chromatography (SEC; Hi Load16/60 Superdex 200) in purification buffer. Clean protein fractions were pooled and snap frozen to be stored at −80 °C. Nup98 in pET28a was purified as described above with the following changes: the buffers used were purification buffer (6 M GuHCl, 50 mM The washes buffer is 6 M Gu H2C, and the elution buffer is 500 mM. GuHCl, 500 mM imidazole pH 8). After induction with IPTG, cells were grown further for 3 h at 30 °C before being collected. Sufficient purity of the protein was achieved with affinity chromatography; consequently, no SEC was performed. The His-tag was still there. TheProtein–Ni was kept at 4 C for phase-separation.
An oil immersed objective was used for the image using the LSM 880 inverted laser scanning confocal microscope. The substrate–Nup98 reaction mixes were transferred into a 12-well silicone chamber (Ibidi) on a 170 ± 5 µm cover slide. Z-stacks were taken around the centres of the phase-separated Nup98 condensates (position with highest diameter) with sequential imaging at 488 and 568. Images were processed using ImageJ2 v.2.9.0/1.53t, MATLAB R2020a and Prism v.9.4.1.
Experimental details for Fig. 4 and Extended Data Fig. 9b–d: CLP-mCherry was assembled with a ratio of 1:100 CA-mCherry (final 0.4 µM) to CA (final 40 µM) by addition of NaCl (final 1 mM) and 15 min incubation at 37 °C. After assembly, the sample was spun down hard (18,000g, 7 min) to separate the assemblies from non-assembled monomer. The pellet was resuspended in assay buffer (50 mM Tris pH 7.5, 150 mM After resuspension the same as the sample volume, followed by a slow spin of the resuspended pellet to remove aggregates. Nup98 condensates were immediately mixed with CA-mCherry assemblies (1 µM monomer concentration) and imaged after around 5 min incubation on the coverslip.
The CA-mCherry and CAhexamer-mCherry (both final 25 M) were part of the experimental details for Extended Data.
Z-slices were chosen to coincide with the Nup98 condensates. Profiles were background subtracted. The first intensity value greater than 5 was the definition of the edge of a condensate. 200 nm was removed from the value to account for the point spread function. The point was defined in the graphs as being 0 m.
Unlabelled CLPs were prepared as described above. A confocal microscope was used in a two step centrifugation protocol. Equal volumes of CLP suspension were mixed with freshly shock-diluted condensates, immediately before to plunge freezing to minimize sample aggregation.
A mixture of Nup98 condensate and capsid was applied onto a glow-discharged quantifoil R2/2 copper grid (Quantifoil Micro Tools). The grid was put through a 5 second test at 15 C with 70% relative humidity and plunged into liquid ethane using a Lecia micro system. The vitrified grids were then stored in liquid nitrogen before cryo-ET imaging.
The grids were imaged on a Talos Arctica electron microscope (Thermo Fisher Scientific) operated at 200 kV acceleration voltage. Cryo-ET data were collected with single-axis tilt on a Falcon III direct electron detector (Thermo Fisher Scientific) in linear mode at a magnification of ×28,000 with a pixel size of 5.23 Å. Tilt series were collected using the dose-symmetric scheme60 from −60 to 60° at 3° intervals using Tomography software (Thermo Fisher Scientific) with the defocus value set to −10 µm. The total dose for each tilt series ranged from 60 to 70 e/2. Before tomogram were reconstructed, images of tilt series were binned two-fold. The IMOD package61 is used to generate three-dimensional reconstructions. The tilted images were aligned with the help of fiducial tracking.
Confocal microscopy images and graphs in Fig. 3 and Extended Data Figs. There are more than two independent experiments with the same results. Three experiments with similar results are represented by the images in figs. There are graphs and images of cryo. 4b–h and Extended Data Fig. 9 show representative data from two independent experiments with similar results.
Cell-based cloning of Nup42 and Nup54 in the pDONR221 Gateway Master Vector for the production of pET28a and pGEX-6P-3 Proteins
Nup42, Nup50, Nup54, Nup62, Nup88, Nup133 and Nup214 in the pDONR221 gateway master vector were purchased from DNASU and cloned into the cell=free expression vector pCellFree_03 (ref. The Gateway method can be used to produce fusion proteins with an N-terminal GFP tag. Nup35, Nup58, Nup98, Nup358 and Pom121 were cloned from complementary DNA (cDNA) prepared from HEK293T cells. Briefly, RNA was extracted using an Isolate II RNA mini kit (Bioline), and cDNA was synthesized using oligo(dT) primers and SuperScript IV (Thermo Fisher Scientific). Target genes were then cloned into pCellFree_03 using the Gibson assembly protocol (New England Biolabs). Nup98 and Pom121 fragments were purchased as gBlocks from IDT and cloned into pCellFree_03. pET28a vectors containing mCherry2-Nup98 fragments, as well as pGEX-6P-3 vectors containing importin-α, TNPO1 and TNPO3 were purchased from GenScript. Cell lines (HEK239T) were only used for cDNA preparation in this study, with gene identity subsequently confirmed by Sanger sequencing.
Importin-α, TNPO1 and TNPO3 were expressed as GST-fusion proteins from E. coli Rosetta2 cells, as described above for importin-β and IBB-mCherry. Cell pellets were then resuspended in 25 mM The tris has a pH of 7.5. 1 mM DTT, 10% glycerol, and 1 cOmplete edt-free Protease Inhibitor (Roche) were lysed. Cell lysates were subjected tocentrifugation for a period of sixty minutes. Clarified lysates were bound to GSH-resin and washed with 25 mM Tris pH 7.5, 150 mM A 1 mM DTT, 10% glycerol, is contained in NaCl. PreScission Protease was used to achieve oncolumn cleavage of the GST-tag. The Cleaved was subjected to the SEC in 50 mM and the equilibrated in 26/600. Tris pH 7.5, 200 mM NaCl, 1 mM DTT, 10% glycerol. Eluted proteins were concentrated, snap frozen and stored at – 80 °C.
To assemble the final cross-linked hexameric CAhexamer-mCherry, CAhexamer and CAhexamer-mCherry were mixed in a 5:1 ratio and assembled by means of the following dialysis steps: twice against 50 mM The tris have pH 8, 1,000 mM. NaCl, 2 mM DTT; twice against 50 mM Tris pH 8, 1,000 mM NaCl; and twice against 50 mM The tris had a pH of 8. NaCl. The reassembled CAhexamer-mCherry was pooled and frozen to 80 C, because it was finally purified and carrying one mCherry.
For assembly of CLPs, untagged CAwt was overproduced in E. coli LOBSTR-RIL(DE3) cells (Kerafast). Cells were resuspended from the lysis buffer. Tris/HCl pH 8.0, 50 mM NaCl, 4 mM TCEP, 2 mM PMSF and Turbo Nuclease). Cells were lysed and cleared at 20,000g for 60 minutes. CA was precipitated from the cleared lysate. The precipitated antibody was collected for 20 minutes. The pellet was resuspended and dialysed overnight against 20 mM, after it was discarded. Tris/HCl pH 8.0 and 1 mM TCEP is a term used to describe something. CA was further purified after injecting on a HiTrapq column and eluting it. Tris/hcl has a pH 8.0 and 100 mM. NaCl, 1 mM TCEP. The columns was regenerated with 50 mM. Tris/HCSl was pH 8.0, with 2.0 M NaCl. TCEP to remove nucleic acid contaminations. The SEC exchanged the eluted CA for 25 mM. Tris/HCl pH 8.0. Fractions were pooled and concentrated to approximately 19.0 mg ml−1.
HIV-1 CA proteins (K158C, A204C, R18G/A204C, N57D/A204C, N74D/A204C, N77V/A204C) were expressed in Escherichia coli C41 or Rosetta2 cells and purified as previously described56. HIV-1 CA K158C was labelled with Alexa Fluor 568-C5-maleimide (AF568, Thermo Fisher Scientific, A20341) and mixed in an approximately 1:200 ratio with unlabelled HIV-1 CA A204C before assembly. CA lattice assembly was carried out as described by Lau et al. 17 with the modification of a 15 min incubation at 37 °C and no overnight incubation. The CA-mCherry, CA-dCherry and CA-dCherry were made up of different substances. Briefly, CAhexamer, CAhexamer-mCherry (pOPT) and CA-mCherry (pET21a) were expressed in E. coli C41 cells. Cells were grown at 37 °C, protein expression was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at an optical density of 0.6, and cell growth was continued overnight at 18 °C. Cells were collected and resuspended in a buffer of 50 mM. Tris pH 8, 50 mM NaCl, 2 mM dithiothreitol (DTT); +cOmplete EDTA-free Protease Inhibitor (Roche)) and lysed by sonication. The lysate was cleared bycentrifugation and the precipitation was carried out on the fraction. After stirring for 30 min at 4 °C, the precipitated material was pelleted (16,000g, 20 min, 4 °C). The pellet of CAhexamer was re-crystalized using 100 mM citric acid pH 4.5, 2 mM DTP and dialysed against the same buffer three times. The precipitated and the soluble fractions were collected for further purification. CAhexamer-mCherry and CA-mCherry could not be purified with citric acid precipitation owing to a possible loss of mCherry fluorescence. The ammonium-sulfate-precipitated pellets were resuspended in 8 ml purification buffer per litre culture, run over a 20 ml Hi-TRAP Q column in purification buffer and eluted in a 1–100% gradient with buffer B (50 mM Tris pH 8, 500 mM It’s NaCl. Protein-containing fractions were pooled.
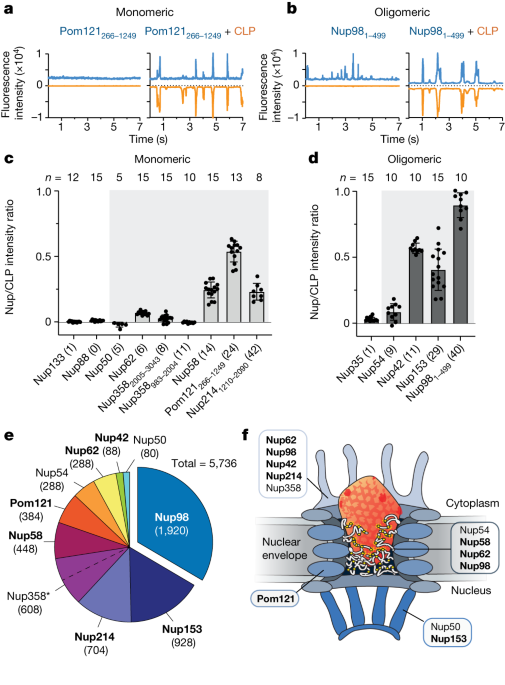
The European Cell Culture Collection had HeLa-K cells that were tested negative for mycoplasma. Cells were grown with a mixture of high-glucose cells and fetal calf semen on 8 well -slides. The cells were treated with a water-soluble fraction in the transport buffer. Permeabilized cells were then put to sleep for 30 min with 40 nM of various anti-Nup133 and EGFP and 40 nm viral capsid spheres. Where indicated, 3 µM of a RanQ69L fragment comprising residues 1–180 and charged with GTP was also added. The samples were then directly scanned with a Leica SP8 confocal laser-scanning microscope (equipped with a ×63 oil objective and HyD GaAsP detectors), using the 488 and 638 nm laser lines sequentially for excitation. Such live scan directly reads the distributions of the analysed probes between bulk buffer and NPCs—although local concentration at NPCs is underestimated because of the diffraction-limited resolution.
A total of 4 µl of purified CA hexamer, CA-N21C/A22C capsid spheres and CLPs at 0.025, 0.1 and 0.25 mg ml−1, respectively, were adsorbed for 1 min on carbon-coated 300-mesh copper grids (EMS) glow-discharged for 1 min at −15 mA using a PELCO easiGlow instrument. Sample droplets were blotted with filter paper held perpendicular to the grids. The grids were then quickly washed three times in 25 μl MilliQ water droplets, followed by staining in 25 μl droplets of 2% uranyl acetate solution for 10 s and again for 1 min, with blotting between droplets. After the final staining, grids were thoroughly blotted with filter paper and allowed to air dry before imaging. Micrographs were collected on the microscope with the Gatan Ultra Scan 2k 2k detector. Representative micrographs were imaged with a defocus range of −1 to −2 μm at nominal magnifications of ×52,000 for CA hexamers or ×15,000 for capsid spheres and CLPs with pixel sizes of 4.15 and 1.25 Å, respectively.
Assembly was performed in a high-salt buffer containing 50 mM At room temperature, a tris/hcl pH 8.0 and 100 M IP6 at a concentration of 10–15 kmJ can be taken for a 24h period. The sample was then dialysed against 50 mM Tris/HCl pH 8.0. The ethamers were isolated by size exclusion chromatography (SEC) using a Superdex 200 increase column. Tris/HCl pH 8.0, 150 mM NaCl, 100 µM IP6. The samples were stored at 80 C for later use. The data was validation by polyacrylamide gel electrophoresis.
The disulfide- CA hexamers were prepared in accordance with the instructions in the ref. 49. The CAP1A/A14C/E45C/W189A/M185A mutants was improved by adding an His14 tag and a bd-SUMO tag. Recombinant expression was in Escherichia coli LOBSTR-RIL(DE3) (Kerafast) cells66. The cultures were stimulated with half a mM. IPTG at 18 °C for 16 h. Cells were resuspended in the lysis buffer. Tris/HCl pH 8.0, 500 mM NaCO2, 20 mM imidazole, 1 mM. TCEP, 2 mM PMSF, are lysed with a high-pressure cell homogenizer. centrifugation cleared the lysate at 8,500g. The fraction was prepared with Ni Sepharose 6 Fast Flow beads. The beads were subsequently washed with wash buffer (50 mM Tris/hcl has a pH 8.0 and 40 mM imidazole. One mM TCEP. Proteins were eluted from the beads by tag cleavage with 2–5 µg ml−1 of bdSenP1 in cleavage buffer (50 mM Tris/HCl pH 8.0, 150 mM The temperature was 4 C for 3 h.
The Supplementary Table 1 has the names of plasmids used for expression. The details of expression and purification were detailed in the references. 21,25,26,50,64).
Assay buffer for nup 98, yeast 116, and human globular flocculositide (FG) particles using tris/HCl pH 7.5, 150 mM NaCl, 1
Partition coefficients were used to divide the raw signal in independent FG particles by the reference areas outside of the region. Plots are shown for representative FG particles (with 4–7 μm diameters). Images were analysed in FIJI 2.9.0 and the exported data were further processed in GraphPad Prism 9.5.1.
The GLFG stock was dissolved in 2 M guanidinium hydrochloride and the other FG stocks in 4 M guanidinium hydrochloride. Assay buffer for human Nup98-FGs and yeast Nup116-FGs was 50 mM Tris/HCl pH 7.5, 150 mM NaCl, 1 mM IP6.
The assays were performed as previously described25,50 with minor modifications. In brief, phase separation was initiated by rapidly diluting a 1 mM FG domain stock with 25 volumes (for GLFG) or 50 volumes (for the others) of assay buffer (50 mM Tris/HCl pH 7.5, 250 mM After a fourfold dilution in the buffer with the indicated fluorescent probes, comes a 1 mM NaChemical. The resulting mixture was placed on collagen-coated μ-slides 18-well (IBIDI) and FG particles were allowed to sediment to the bottom of the slides for 1 h before confocal scans were taken.
Source: HIV-1 capsids enter the FG phase of nuclear pores like a transport receptor
A fully grown CD1 mouse model imaged by a biosensor in M2 medium supplemented with dibutyryl cyclic AMP
Mouse oocytes were obtained from ovaries of 9-week-old CD1 mice which were maintained in a specific pathogen-free environment according to the Federation of European Laboratory Animal Science Association guidelines and recommendations, as previously described53. Fully grown oocytes were kept arrested in prophase in homemade M2 medium supplemented with 250 μM dibutyryl cyclic AMP under paraffin oil (NidaCon) at 37 °C. The capsid spheres were injected into the nucleus of oocytes. About 25 min after the injection, oocytes were imaged.
The biosensor tips were pre-incubated in the buffer for 10 minutes. Tris/HCl pH 8.0, 300 mM NaCl, 0.1%, bovine albumin, 0.05%, and Tween-20. After dipping the tips into the BLI buffer, the ligands were conjugated to a thick layer of 1 nm for 30–45 s. Association was recorded for 180–300 s, followed by a 200–300 s dissociation step in wells containing BLI buffer. The instrument had a fortebio OctetRED96. There was a reference sensor included that was used to subtract noise. Data were normalized to the baseline step and aligned to the dissociation start point using the Octet data analysis software. Data was plotted using a data analysis program.

