Single-cell histones are used for embryo tracing
Single-embryo RNA-seq Library Preparation with a Smart-Seq3 Protocol 51,52
scRNA-seq and single-embryo RNA-seq library preparation were performed using a modified Smart-seq3 protocol51,52. Tyrode treated with a solution called Sigma to remove the zona pellucida. There were single cells or embryos isolated at the blastocyst stage after being mouth-pipeted into lysis buffer. There was a lys buffer consisting of 0.15% VWR Life Science, 5% PEG8000, 0,5 M oligo-dT, and 0.5 mM dNTPs. After the procedure, the lysis tubes were put through a process to make sure the lysis buffer was found. Tubes of sorted cells were denatured at 72 °C for 10 min, followed by the addition of the reverse transcription mix. The reagent concentrations were as follows: 25 mM The tris-hcl is composed of 30 mM NaCl, 2.5 mM Grp (Thermo Fisher Scientific), and 8 mM. DTT ((Thermo Fisher Scientific), 0.25 U μl–1 RNase inhibitor, 2 μM TSO (5′-AAGCAGTGGTATCAACGCAGAGTACATG(r)G(r)G(+)-3′) and 2 U μl–1 There is a reverse transcriptase called Maxima H Minus byThermo Fisher Scientific. Reverse transcription and template switching were carried out at 42 °C for 90 min followed by 10 cycles of 50 °C for 2 min and 42 °C for 2 min. After 5 minutes, the reaction was stopped by initument at 85 C. Indicated volumes of PCR master mix were dispensed, which contained 1× KAPA HiFi PCR buffer (Roche), 0.3 mM dNTPs each (Roche), 0.5 mM MgCl2 (Roche), 0.6 mM P2 primer (5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-3′) and 0.2 μM IS primer (5′-AAGCAGTGGTATCAACGCAGAGT-3′) and 0.02 U μl−1 KAPA HiFi DNA polymerase (Roche). Pre-amplification was performed as follows: 3 min at 98 °C for initial denaturation, 16 cycles of 20 s at 98 °C, 30 s at 65°C, and 5 min at 72 °C. Final elongation was performed for 5 min at 72 °C. After PCR, samples were pooled and purified using a TIANquick Mini Purification kit (Tiangen) and 0.8× AMPure XP beads (Beckmann).
The sgRNAs targeting the promoter of each of the candidate totipotency-related TFs were synthesized and inserted into a CROP-opti vector separately (Addgene, 106280) (Supplementary Table 9). The libraries included candidate sgRNAs and positive control sgRNAs. The supernatant was captured for 18 h and then used to remove cell debris. The mouse ES cells were infected (8 μg ml–1 polybrene) with various titres of lentivirus to achieve different multiplicity of infection values. At 24 h after transduction, new culture medium with 2 μg ml–1 puromycin was added for 48 h for selection. Cells were collected after they were transduction and selection. The cell pellets were preserved at 80 C after being fixed at 4 C for 10 minutes. The process of single-cell ribonucleic acid sol-sq for capturing both nr and sgRNAs was done as per the method described54.
The enrichment analysis is in fig. The observed and expected probability were calculated using the length of the classifier bins and the length of the mouse genome, respectively. 75 transposable elements were found to be highly enriched in 2,583 totipotency-related bins. For the annotation of chromatin states of the enriched 75 transposable elements in Fig. 4g, we calculated the percentage of transposable element copies that were defined as promoters, enhancers, gene bodies, heterochromatin or quiescent/low regions for each synthetic cell. To quantify expression levels of these transposable elements, we mapped raw scRNA-seq reads to the mm10 genome using Hisat2 (v.2.2.1)69 and filtered out mapped reads with MAPQ less than 10 with Samtools (v.1.9). We put the numbers of TPM together using the location of the repeats downloaded from the UCSC Genome browser.
Source: Genome-coverage single-cell histone modifications for embryo lineage tracing
Detailed Analysis of the 2 Cell Phase of GSE8218567 using Homer HiC, Seurat, UMAP, and FindMakers
The allvalidPairs matrix for the late 2cell stage was downloaded from the GEO database (accession number GSE82185)67. To identify interactions, we used the analyzeHiC function of Homer (v.4.11)68 at 50 kb resolution and plotted interactions with Python (v.3.9.7).
The promoter pairs for which H3K4Me3 peaks or H3K27ac peaks fell into copies of the MERVL elements were chosen because they function as enhancers. In addition, we identified 332 enhancers and 866 enhancers in which MERVL elements functioned as enhancers.
In Extended Data Fig. 10b, for each chromatin-state labelling, after TF–IDF normalization, SVD dimensionality reduction and cluster finding (as described above), we used the FindMakers function in Seurat (v.4) to find differential bins between cells at the 2cell and 8cell stage. We looked at the number of differential bins that were already annotated as corresponding chromatin states in each synthetic cell to calculate the percentage of differential bins that were already established.
Seurat’s SCTransform function normalized each sample and the samples were integrated using Seurat’s anchoring and integration functions. The first fifteen components of PCA were used to generate a shared-nearest-neighbours graph. The Louvain algorithm, which has a resolution of 0.2, was used to determine the ratio of nuclei to each other. UMAP was performed using the first 15 principal components and 30 nearest neighbours. The FindConservedMarkers function applied markers to each cluster to be annotated for cell type.
To integrate the six histone modification profiles, we used the multivariate HMM introduced in ChromHMM61. We binarized all .bam files for each synthetic cell using the binarizeBam function of ChromHMM with default parameters. We used the Learn Model function to learn the 12 states of each synthetic cell. To reduce noise and mitochondrial interference, all reads from mitochondrial DNA are filtered out. We annotated each state on the basis of what we found, and there were three steps that needed to be taken: 1) filtering out low genome coverage states, 2) defining hidden states based on histone modifications, and 3) aligning the annotations on the basis of overlap. Finally, we labelled the 12 states as multivalent (all histone modifications), weak promoters (H3K4me3), strong promoters (H3K4me3 and H3K27ac), weak enhancers (H3K4me1), strong enhancers (H3K4me1 and H3K27ac), poised gene bodies (H3K36me3 and repressive histone modifications), active gene bodies (H3K36me3 and active histone modifications), polycomb-protein-associated heterochromatin (only H3K27me3), H3K9me3-associated heterochromatin (only H3K9me3), heterochromatin (H3K27me3 and H3K9me3), and quiescent/low.
Having obtained 155 RNA synthetic cells interpolated with six histone-modification profiles, we performed hierarchical clustering with RNA synthetic cells on the basis of multimodal histone modifications. Two clusters for the 2cell stage are similar to four clusters for the 4cell stage, and so on. Next, we aggregated histone-modification profiles of cells in the same cluster, which led to 90 synthetic single cells with joint profiles of 6 histone modifications. We normalized cell numbers and non-duplicated reads to reduce the effects from sequencing depth.
We integrated H3K27ac CoTACIT profiles with gene expression in the same way as described in step (2). As H3K27ac, H3K27me3 and H3K9me3 profiles were experimentally linked, we directly transferred corresponding H3K27me3 and H3K9me3 profiles to the linked RNA synthetic cells.
We ordered scRNA-seq cells along the developmental trajectory using Monocle3 (ref. 60) and merged five adjacent single cells along pseudotime into one RNA synthetic cell.
To calculate the genome coverage at each developmental stage, we first called peaks for aggregated .bam files of each histone modification. We used MACS2 to call peaks with parameters of ‘–nolambda–nomodel -q 0.05–broad’. The amount of genome intervals that overlap with peaks at a specific stage was calculated using the binned genomics and mapped to the peaks at that stage. The genome was first put in a bin of 200 bpm and the histone modification signals in the bin were used to see how much coverage the genome had. The percentage of covered bins is determined by the coverage of each single cell.
The multiBigwigSummary function in deepTools gave us normalized mean scores in 5-kb bins of the genome for correlation analysis. The Spearman correlation or Pearson correlation was calculated between replicates and plotted using the plotCorrelation function.
In Vitro Incubation of Blast Cysts with Paraformaldehyde for the Initrogen-Dilution Process
When the embryos were injected, they were fixed with 4% paraformaldehyde for 10–15 minutes. The embryo was incubated with SOX2 and CDX2 at 4 C overnight. After 3 wash-ups, the samples were washed with PBS for a second time to be used in the invitrogen-dilution process. After 5 min at room temperature, blastocysts were washed and put into PBS with 600 nM DAPI solution. Images were acquired using a confocal microscope.
Next, cells were incubated with specific antibody in 100 μl antibody buffer (20 mM HEPES pH 7.5, 150 mM 3 mM spermidine, 2 mM EDTA, 0.05% digitonin, 1 mM sodium butyrate and 1 mM PMSF are at 4C for 2–1 h. The cells were washed again with a buffer of at least 20 mM HEPES 7.5. 10 mM sodium butyrate and 1 mM PMSF are suspended with a high-salt buffer. Nacl, 0.10% digitonin, 0.05% TX-100, 1 cocktail, 10 mM sodium butyrate, and 1 mM PMSF are contained. The PAT expression, purification and assembly procedures were performed as per previously described guidelines19. Cells were rotated at 4 °C for 1 h to enable complete binding of PAT to antibodies and then washed twice with 180 μl high-salt Dig-wash buffer to remove free PAT–MEA/B. Tagmentation was reactivated by suspending cells with 10 μl cold reaction buffer (10 mM TAPS-NaOH pH 8.3, 5 mM MgCl2, 1× cocktail, 10 mM sodium butyrate and 1 mM PMSF) and incubated at 37 °C for 1 h in a PCR cycler. The reaction was stopped by adding 10 μl 40 mM A 96-well plate was used to pick and place single cells into after being washed twice with 1% BSA–PBS. The 96-well plates were pre-rinsed with 1% BSA–PBS to avoid loss of DNA fragments, and 2 μl lysis buffer (10 mM Each well received Tris-HCl pH 9.0, 0.05% SDS and 0.1 g of 1Proteinase K. For each well, samples were covered with 5 μl mineral oil (Sigma) and incubated at 55 °C for 15 min to release DNA fragments. Next, 0.5 μl of 10 mM PMSF was added to each well to deactivate protease K, and 1 μl of 0.9% Triton X-100 was added to quench SDS in the reaction. Finally, 17 μl PCR mix (0.2 μl The KAPA High-GC buffer, 0.25 l 10 mM dNTP mix, and 25 mM MgCl2 were added to each well. The Nextera i5 and 10 mM i7 index primer were the subject of a Supplementary Table 1. PCR enrichment was performed in a thermal cycler with the following program: 1 cycle of 72 °C for 5 min; 1 cycle of 95 °C for 3 min; 11 cycles of 98 °C for 20 s, 65 °C for 30 s, 72 °C for 1 min; 1 cycle of 72 °C for 5 min; and hold at 4 °C. The library was purified with 1× AMPure XP beads (Beckman) once, and 200–1,000 bp fragments were selected with 0.5× + 0.5× The beads are from AMPure. The libraries were sequenced with paired-end 150-bp reads on a NovaSeq 6000 platform (Illumina).
For siRNA knockdown, isolated zygotes were microinjected with sets of three siRNAs against targets (20 μM in total) or with non-target control (NC, 20 μM in total). The following siRNAs were used. Hippobio ordered the siRNAs. The injected embryos were transferred to KSOMaa medium, and then the droplets were covered in mineral oil and cultured in a tissue incubator. The embryo was collected at the 8cell stage, and single-embryo had been used to confirm KD or marker genes.
The following antibodies were used for TACIT (catalogue and lot numbers provided after the supplier name): H3K4me1 (1:50; Abcam, ab8895, GR3369516-1); H3K4me3 (1:200; Millipore, 04-745, 3243412); H3K27ac (1:500; Diagenode, C15410196, A1723-0041D); H3K36me3 (1:200; Active Motif, 61101, 06221007); H3K27me3 (1:200; Millipore, 07-449, 3146226); H3K9me3 (1:200; Active Motif, 39161, 30220003); and H2A.Z (1:200, Abcam, ab4174, GR279096-1). Donkey anti-rabbit-Alexa 488 (1:500; Invitrogen, A32790) and donkey anti-rabbit-Alexa 555 (1:500, Invitrogen, A31572) were used as secondary antibodies. Antibodies used in immunofluorescence staining included SOX2 (1:200; Active Motif, 39843, 2226414) and CDX2 (1:200, BioGenex, MU392A-UC, MU392A0516D).
Wild-type V6.5 mouse ES cells were cultured at 37 °C with 5% CO2 and were maintained on 0.1% gelatin-coated plates in high-glucose DMEM culture medium containing 15% fetal bovine serum (Invitrogen), 1% penicillin–streptomycin (Hyclone), 1% MEM nonessential amino acids (Cellgro), 1% Glutamax (Gibco), 1% nucleoside (Millipore), 0.1 mM 2-mercaptoethanol (Sigma) and 1,000 U ml–1 recombinant leukaemia inhibitory factor (Millipore).
Source: Genome-coverage single-cell histone modifications for embryo lineage tracing
C57BL/6 Mice. Experiments and Data Handling with GraphPad Prism Software (v4.1.1) and Extended Data Analysis (Extended Abstract)
To collect zygotes, a cumulus mass containing several zygotes surrounded by follicular cells was transferred to 1× hyaluronidase solution (Sigma) and incubated at 37 °C for a few minutes. The zygotes were then transferred to M2 medium and their zona pellucida was gently removed by treating with pre-warmed Tyrode’s acidic solution (Sigma) for several minutes. The second polar bodies of zygotes were manually removed with a very fine glass needle.
Aged C57BL/6 mice (16–21 months old) were obtained from the National Institute on Aging rodent colony. Young C57BL/6 mice (3 months old) were obtained from Jackson Laboratories. All experiments used male mice. All mice were kept on a 12 h–12 h light–dark cycle and provided ad libitum access to food and water. All animal care and procedures complied with the Animal Welfare Act and were in accordance with institutional guidelines and approved by the Veterans Affairs Palo Alto Committee on Animal Research and the institutional administrative panel of laboratory animal care at Stanford University.
Data handling and statistical analyses were performed using R studio (v4.1.1) and GraphPad Prism Software unless stated otherwise. All statistical analyses comparing measurements between two groups were carried out using unpaired two-tailed Student’s t-tests. Post hoc tests for multiple comparisons and one- way ANOVA tests were used in all statistical analyses. A P value of 0.05 is significant. When possible, the procedures were carried out in alternating fashion in order to avoid temporal and technical biases. There is data in figs. 1b–d,h–o, 2a,b,f–j, 3e–n, 4f–i and 5j–l and Extended Data Figs. 2a, 3f–i, 7a–c,h,i and 8h–k were successfully replicated in at least two independent experiments.
In fear conditioning tests, mice were trained to associate cage context and an audiovisual cue with a foot shock. On the first day of training, mice were put in a cage and exposed to two periods of light and tone, followed by a shock with a 180-s interval. On day two, mice were re-exposed to the same cage context and recording freezing behavior was done using a FreezeScan tracking system. mice were placed in a novel context that contained different smell, floor texture and chambers and were exposed to the same light and tone from day 1 after 2 min of exploration. Freezing behaviour was recorded for 1.5–6 min following the cue using the FreezeScan tracking system (Cleversys).
The Y maze is made up of 3 white, opaque plastic arms at 120 angles from each other. At the beginning of the trials, mice were placed in the end of 1 arm and allowed to freely explore all 3 arms for 5 min. A definition of an arm entry is having all four limbs in an arm. The maze was cleaned with 70% ethanol between animals and before the first animal to eliminate traces of odour. The number of arm entries and triads (a set of consecutive arm entries) were recorded. By dividing the number of combinations by 100, the spontaneous alternation was calculated.
Pseudobulk counts for Metascape43 using the Aggregate Expression function. Data normalization, differentiation, and correction using the Benjamini-Hochberg procedure
Pseudobulk counts were derived by aggregating raw counts for each sample using Seurat’s AggregateExpression function. The default parameters were used to perform the bulk data normalization and differential expression across the groups. P value correction was carried out using the Benjamini–Hochberg procedure (FDR = 0.05) for each comparison. There are genes with FDR that were used for the Metascape43 analysis.
The M musculus reference genome was aligned with the CellRanger software in order to obtain Gene counts. Ambient RNA was removed from each sample using SoupX (v1.6.2) and droplets containing multiple nuclei were filtered out using DoubletFinder (v2.0.4). We used Seurat (v4.1.1) to further exclude cells with fewer than 200 or more than 5,000 features and cells with more than 10% mitochondrial genes. In total, 69,250 nuclei remained and were used for further analysis (Supplementary Fig. 3a–d.
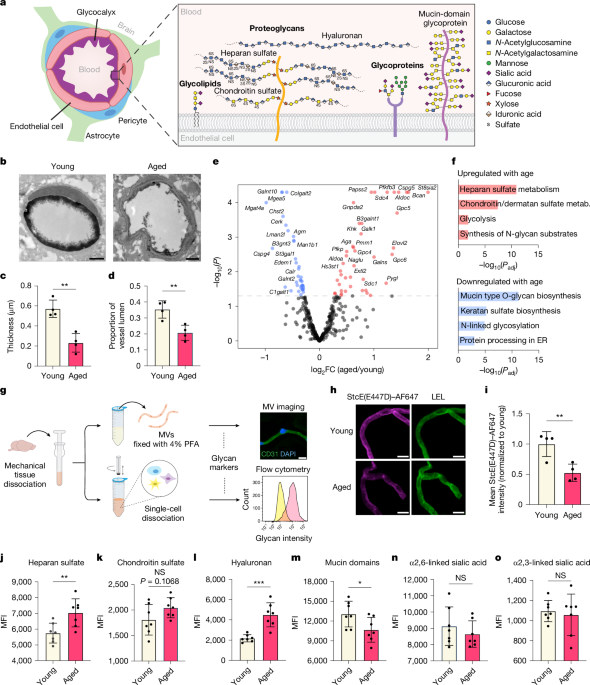
Source: Glycocalyx dysregulation impairs blood–brain barrier in ageing and disease
Mechanical Dissociation of Microvessels from Mice for Brain Tissue Enhancement using FACS Buffer (PBS) Gelentriches
Some modifications were made to the way in which microvessels from young (3-month- old) and aged (21-month-old) mice were isolated. The brains of the mice were retrieved in PBS supplemented with 1% bovine albumin and 1 cOmplete protease Inhibitor cocktail and ice was used to dispose of them. The olfactory bulb was discarded, and meningeal vessels were removed by gentle rolling on blotting paper. Brains were finely ground using a razor blade and then put into a container. Dounce (Wheaton) in 1% BSA-PBS with 1× protease inhibitor. The homogenate was centrifuged in 27% (wt/vol) 70 kDa dextran (Sigma) in HBSS at 4,400g for 25 min. Myelin and parenchymal cell layers were removed. Pelleted microvessels were deposited on a pre-wet 40-μm strainer, washed with PBS, and mechanically dissociated into single cells as previously described26. For enzymatic dissociation of brain tissue, previously published protocols were used12,13. Pelleted cells were suspended in FACS buffer (1% BSA in PBS) and stained on ice for 30 min with the following antibodies: rat anti-CD31-PE/CF594 (1:100, BD, 563616), rat anti-CD45-PE/Cy7 (1:200, Biolegend, 103114), mouse anti-heparan sulfate (1:100, Amsbio, clone 10E4, 370255-1), mouse anti-chondroitin sulfate (1:100, Sigma, clone CD-56, C8035), biotinylated HABP (1:150, Amsbio, AMS.HKD-BC41), fluorescein-conjugated SNA (1:300, Vector Labs, FL-1301-2), biotinylated MAAII (1:300, Vector Labs, B-1265-1), and StcE(E447D)–AF647 (5 μg ml−1). The cells were washed with the FACS buffer and secondary immunizations were carried out on ice for 20 min. Live cells were found using the viability dye, sytox blue. Data was analysed using FlowJo software and a flow cytometry analyzer was used.
It was a gift to produce thePHP.V1 from V. Gradinaru. Triple transfection of HEK293T cells was performed on cells with a high concentration of Glutamax. 12 h postfection, a new warm medium was replaced. Medium was collected 72 h post-transfection. Fresh, warm medium and cells 120 h post-fection were added together. The cells and medium were placed at room temperature for 15 minutes. Supernatant was collected in a separate bottle and combined with 40% (wt/vol) Before transferring to 4 C, the final concentration is placed on ice for 2 hours. The cell pellet was resuspended in a buffer containing salt-active nuclease, then incubated at 37 C and transferred to 4 C overnight. PEG medium was centrifuged at 4,000g for 30 min at 4 °C. Supernatant was thrown out after centrifugation. PEG pellet was resuspended in SAN + SAN buffer, combined with the previous fraction, and incubated at 37 °C for an additional 30 min. lysate was loaded on to an iodixanol gradient using 15%, 25%, 40% and 60% fractions. Gradients were transferred to an ultracentrifuge (Beckman Coulter) using a Type 70 Ti rotor set at 350,000g for 2 h and 25 min at 18 °C. AAV particles were collected from the 40/60% interface, washed in PBS, and concentrated using an Amicon Ultra-15 (Millipore Sigma) filter device with a 100 kDa cutoff. AAV titration was performed using the AAVpro Titration Kit (for Real Time PCR) Ver.2 (Takara Bio). At an average of 8 1011. viral genomes per mouse, AAVs were injected.
The Ple261 MiniPromoter was a gift from E. Simpson. A cis rAAV genome plasmid with AAV2 inverted terminal repeats was utilized for cloning of a sCLDN5 and EGFP reporter using restriction enzymes and In-Fusion Snap Assembly (Takara Bio). To knock down C1galt1 in brain endothelial cells, de novo predictions of small interfering RNA (siRNA) guides targeting C1galt1 were generated using the DSIR algorithm39 and subsequently filtered using ‘Sensor rules’ to select for sequences with highly favourable small hairpin RNA (shRNA) features40,41. Three de novo 97-mer miR-E shRNA genomes were created and inserted into the pAAV-sclDN5-EGFP using restriction enzyme cloning. The cloned P2A-C1GALT1 and P2A-B3GNT3 were used to create pA.
Brain microvessels were isolated and lysed in 1× RIPA buffer with 1× cOmplete protease inhibitor cocktail (Sigma) on ice. Lysates were centrifuged at 13,000g for 15 min at 4 °C, and supernatant protein concentration was measured by microBCA (Pierce). The samples were reduced and alkylated by TCEP and then the purification using SP3 was done. The samples were eluted after being desalted for 3 h at 37 C and with 200 ng trypsin/LicsC in the stomach. Samples were acidified to 0.1% formic acid and filtered before loading onto the nanoLC system. One microgram protein was injected for all samples. LC–MS/MS analysis was performed on the TimsTOF Pro (Bruker Daltonics) coupled with the NanoElute system (Bruker Daltonics) with solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile). Tryptic peptides were loaded first on the trapping column Waters ACQUITY UPLC M-Class Symmetry C18 Trap Column, 100 A, 5 μm, 180 μm × 20 mm, and eluted with analytical column, IonOpticks Aurora Elite CSI 15 × 75 C18 UHPLC column. Elution ranged from zero to 5% B, 9 to 12% B, 9.1 to 12% B, and 27 to 30% B, with a flow rate of 0.35 l min1 from 0 to 9 min. Eluted peptides were measured in diaPASEF mode with base method m/z range 100–1,700 and 1/k0 range of 0.85–1.30 V s cm−2. The source parameters were 1,400 V for capillary voltage, 3.0 l min−1 for dry gas, and 180 °C for dry temperature using Captive Spray (Bruker Daltonics). Collision energies (27 eV and 45 eV) were allocated for 1/K0 = 0.85 V s cm−2 and 1/K0 = 1.30 V s cm−2, respectively. Data were processed using Spectronaut (Biognosys AG, v19.1) for directDIA search with Swiss-Prot Mouse database downloaded on 3 March 2023. Default settings were used with a slight modification of minimum peptide length 6. The candidates were narrowed down using the Q and log2 ratios.
For confocal imaging analysis, bEnd.3 cells were plated on round coverslips (EMS, 72196-12) in a 24-well plate and treated with 5 nM StcE for 16 h at 37 °C. Cells were fixed in 4% PFA for 15 min, blocked in 3% normal donkey serum with 0.3% Triton X-100 in PBS for 1 h at room temperature, and incubated at room temperature in blocking solution with the following primary antibodies for 1.5 h: goat anti-CD31 (1:100, R&D, AF3628), mouse anti-ZO1 (1:100, Thermo Fisher Scientific, 33–9100), rabbit anti-CAV1 (1:100, Cell Signaling Technologies; 3267S) and mouse anti-CLTC (1:100, Thermo Fisher Scientific, MA1-065). Cells were subsequently washed 3 times with PBS, stained with the appropriate Alexa Fluor-conjugated secondary antibodies (1:250, Thermo Fisher Scientific) for 1 h at room temperature, washed 3 times again, mounted, and coverslipped with Vectashield Hardset Antifade Mounting Medium with DAPI (Vector Labs, H-1500-10) or ProLong Gold Antifade Mountant (Thermo Fisher Scientific, P36934). Imaging was performed on a confocal laser-scanning microscope (Zeiss LSM880), and images were analysed using ImageJ.
The mouse brain endothelial cell line bEnd.3 (ATCC, CRL-2299) was cultured in high-glucose DMEM (Thermo Fisher Scientific, 10567022) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and maintained in a humidified incubator containing 5% CO2 at 37 °C. For bulk RNA-seq analysis, bEnd.3 cells were grown in 6-well plates and treated with 5 nM StcE for 16 h at 37 °C. Cells were lysed and collected into RNAse-free Eppendorf tubes for total RNA extraction using the RNeasy Plus Micro kit (Qiagen, 74034). RNA quantity and quality were assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies). All samples passed a high quality control threshold (RNA integrity number ≥9.7) and proceeded to cDNA library preparation by Novogene. The NovaSeq 6000 had a 2 150 bp depth. There wererimmed reads that were aligned to the reference genome. The analysis and visualization of differential genes was done using DESeq2 (v1.32). Genes with a Padj < 0.05 were used for GO biological pathway enrichment analysis.
Several previously published datasets demonstrate robust brain endothelial cell enrichment and were chosen for the analysis. We looked for genes that are linked to glycosylation using the Encyclopedia of Genes and Genomes. Most glycoproteins were excluded due to the enormous variety of members in this family which possess biological functions not directly relevant to glycosylation. In each dataset, significant up and downregulated glycosylation-related genes were used for the analysis of the reactionome pathway.
LC–MS/MS analysis was performed on a Q Exactive HF-X (Thermo Fisher Scientific) with an UltiMate 3000 RSLCnano system (Thermo Fisher Scientific). There was a 75 m capillary column filled with 40 cm of ReproSil- Pur 120 C18-AQ 1.9 m resin. A separation was achieved with a flow rate of 300 nl min 1 and a 120 min gradient that has a mixture of A + B for 18 min, A + B for 72 min, and A + B for 15 min. The full data-dependent mode scans were obtained with an automatic gain control target of 3 106 and a maximum injection time of 20 ms. MS2 scans were acquired with the following parameters: resolution of 15,000, AGC target of 1 × 105, maximum IT of 54 ms, loop count 15, TopN 15, isolation window 1.4 m/z, fixed first mass 100.0 m/z, normalized collision energy (NCE) 28 units, charge exclusion of unassigned, 1, 6–8 and >8, peptide match preferred, exclude isotopes on, and fragmented m/z values were dynamically excluded from further selection for a period of 45 s. Raw data were processed and analysed using MaxQuant and Perseus36. In brief, peptide spectral matches were made against a target-decoy Mus musculus reference proteome database downloaded from Uniprot. Methionine oxidation and N-terminal acetylation were specified as variable modifications, and carbamidomethylation of cysteines was specified as a fixed modification. Precursor ion search tolerance of 20 ppm and product ion mass tolerance of 20 ppm were used for searches. Both unique and razor peptides were used for quantification. The results were negative for a 1% false discovery rate. The minimum Ratio count for the MaxLF Q37 was set to 1. The missing values were imputed from a normal distribution with width of 0.25 and the log2transformed values from a quantitative comparative analysis. The principal component analysis was performed using the Benjamini–Hochberg FDR. GO term enrichments were performed using DAVID38 with the M. musculus proteome as a background.
Plasma cytokine measurement was performed using the Luminex assay at the Human Immune Monitoring Center at Stanford University. The mouse 48-plex Procarta kit was supplied by the manufacturer. The samples were run in singlet on a 96-well plate, along with standard curve and quality control samples. Custom Assay Chex control beads (Radix BioSolutions) were added to all wells to assess nonspecific binding.
All recombinant StcE proteins were produced as described22,23. At least 7 times the manufacturer’s instructions were followed when running the proteins through the high- capacity endotoxin removal columns. Endotoxin levels were tested using HEK-Blue lipopolysaccharide (LPS) Detection Kit 2 (InvivoGen) according to manufacturer recommendations. Mice were injected retro-orbitally with 0.25 mg kg−1 StcE once a day for 2 days before perfusion with ice-cold PBS. Cerebral bleeding was visualized by eye post-perfusion and by H&E staining. For H&E staining, hemibrains and peripheral organs were formalin-fixed and paraffin embedded (FFPE) and cut into 5-μm-thick sagittal sections mounted on slides. Sections were deparaffinized in xylene (3 times, 3 min), hydrated in a series of graded alcohols (2× 100%, 1× 95%, 1× 80%; 3 min each), and stained with Richard Allan haematoxylin (4 min; followed by 30 s in 4% acetic acid in water and dipping in 0.3% ammonia water) and eosin with phloxine (30 s). Sections were then dehydrated (10 dips in 95% ethanol followed by 2× 1 min in 100% ethanol), cleared in xylene (3 times, 1 min), and coverslipped prior to imaging on a wide-field microscope (Zeiss AxioImager).
Mice were anaesthetized and injected retro-orbitally with Sulfo-NHS-biotin (Thermo Fisher Scientific, 21335) at 0.25 mg g−1 body weight. After 5min the tracer was allowed to circulate. Hemibrains were post-fixed in 4% PFA overnight at 4 °C, cryopreserved in 30% sucrose, and sagittally sectioned into 40-μm slices. Sections were blocked and co-stained with CD31 and the appropriate secondary antibody as described earlier and Alexa Fluor 647-conjugated streptavidin (1:1,000, Thermo Fisher Scientific). Images were taken on a confocal laser-scanning microscope (Zeiss LSM880) and analysed using ImageJ software. Multicoloured gradient images were generated using the fire LUT in ImageJ. Permeability index of vessels was determined as the area occupied by tracer divided by the vessel area.
The mice were euthanized with 2.5%. Avertin and transcardially perfused via peristaltic pump at 2 ml min−1 with the following ice-cold solutions: 8 ml PBS, 10 ml of 5 μg ml−1 of StcE(E447D)–AF647 or SNA–Cy3 (Vector Labs, CL-1303-1), and 8 ml of 4% PFA. The mice were euthanized with 2.5%. Unless noted otherwise, avertin and manually perfused with PBS. Tissues were extracted and fixed in 4% PFA at 4 °C overnight before preservation in 30% sucrose in PBS. Tissues were sectioned into 40 μm slices using a microtome (Leica). Slices were subsequently blocked in 3% normal donkey serum with 0.3% Triton X-100 in TBS-T for 1.5 h at room temperature and incubated at 4 °C overnight with the following primary antibodies: goat anti-CD31 (1:100, R&D, AF3628), goat anti-Iba1 (1:100, Abcam, ab5076), rat anti-CD68 (1:100, Bio-Rad, MCA1957), goat anti-collagen type IV (1:100, Sigma, AB769), rat anti-NID1 (1:100, Thermo Fisher Scientific, MA1-06501), rabbit anti-CLDN5 (1:100, Thermo Fisher Scientific, 34–1600), mouse anti-ZO1 (1:100, Thermo Fisher Scientific, 33–9100), goat anti-albumin (1:100, Thermo Fisher Scientific, A90-134A), goat anti-PODXL (1:100, R&D Systems, AF1556). The following day, slices were washed three times with TBS-T, stained with the appropriate Alexa Fluor-conjugated secondary antibodies (1:250, Thermo Fisher Scientific) or Alexa Fluor-conjugated streptavidin (1:1,000, Thermo Fisher Scientific) for 2 h at room temperature, washed three times again, mounted, and coverslipped with Vectashield Hardset Antifade Mounting Medium with DAPI (Vector Labs, H-1500-10). A confocal laser scanning microscope was used to acquire and analyse images. Luminal vascular coverage was calculated as vessel (CD31+ or COL4A+) area occupied by the marker of interest divided by total vessel area. The MFI was calculated using a CD31+ mask.
The brain tissues obtained from the post mortem frozen state were given the go-ahead by the Stanford Institutional Review Board. Autopsies were performed no more than 12 h after death, and all samples used in this study were stored at −80 °C until the time of processing. The group characteristics are summarized in Supplementary Data 2. Individuals in the Alzheimer’s disease group were both clinically diagnosed and pathologically determined to exhibit Alzheimer’s disease brain hallmarks including β-amyloid and tau pathophysiology.
For BBB ultrastructural analysis, mice were injected retro-orbitally with 0.3 ml of 0.5 mg g−1 of HRP type II in PBS (Sigma, P8250). After 30 min, brains were dissected and fixed in 0.1 M sodium cacodylate buffer (EMS) with 5% glutaraldehyde (EMS) and 4% PFA (EMS) for 1 h at room temperature and then for 16 h in 4% PFA/0.1 M cacodylate at 4 °C. The tissue was washed overnight and then sliced using a matrix into 1-mm-thick sections. Cortical punches (0.5–1 MM3) were cut and brought to the lab where they were put into a jar of 3,3′diaminobenzidine with hydrogen peroxide and put into a chamber for 45 min. Tissues were washed with a 2% osmium tetroxide stain after it was fixed, 2.5% potassium ferrocyanide stain after it was fixed and 1% uranyl acetate stain after it was stained. They were embedded inEMS with the samples being dehydrated in a ascending alcohol gradient. Eighty-nanometre sections were cut using a Leica UC7 ultramicrotome (Leica Microsystems) and collected on formvar-coated 100-mesh copper grids. The grids were stained by the uranyl and lead citrates. Sections were imaged using a Tecnai 12 120 kV TEM (FEI), and data were recorded using a Rio16 CMOS camera with GWS software (Gatan). The assessment of tight junctions in the images was performed in a blinded manner.

