Clinical coding of BRCA2 has been based on genome editing
Using genomic editing to predict the effect of BRCA2 variants in a genome-wide study: A computational approach for the relevance of the missense variant
The prediction of the genome-wide variant effect using deep learning scores was developed by Rentzsch. Genome Med. 13, 31 (2021).
Guidugli, L. et al. Computational and functional approaches were used to assess the clinical relevance of the missense variant. Am. It was J. Hum. Genet. There were 104, 233, and 258 in this year.
Tiberti, M. et al. MutateX: an automated pipeline for in silico saturation mutagenesis of protein structures and structural ensembles. Brief. bbac074 (22) is a Bioinformatics 23.
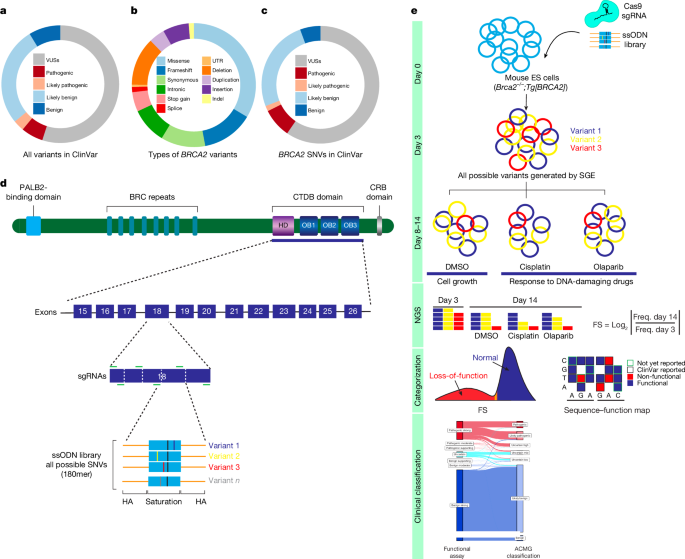
Source: Saturation genome editing-based clinical classification of BRCA2 variants
Automatic interpretation of human genetic variation by the ACMG/AMP framework in genomics and clinical data. Am. J. Hum. Genet. 83, 3861-3867 (2023)
de Bruijn and I. Analysis and visualization of longitudinal genomic and clinical data from the AACR Project GENIE Biopharma Collaborative in cBioPortal. Cancer Res. 83, 3861–3867 (2023).
Sorrentino, E. et al. VarSome is in an existing bioinformatic pipeline, which can be used for automated ACMG interpretation. Eur. Rev. Med. Pharmacol. Sci. 25, 1–6 (2021).
Walker, L. C. et al. Evidence relating to predicted and observed impact on splicing was captured by the ACMG/AMP framework. Am. J. Hum. Genet. There were 105, 104, and 106 in the year 2023.
Easton, D. F. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am. J. Hum. Genet. 81, 822–822.
Cooper and Shendure used a lot of data to find disease-causal variations. Nat. Rev. Genet. It was HRC on 12th of December 2011.
Tabet, D., Parikh, V., Mali, P., Roth, F. P. & Claussnitzer, M. Scalable functional assays for the interpretation of human genetic variation. Rev. Genet. 56, 19.1–19.25 (2022).
Findlay, G. M., Boyle, E. A., Hause, R. J., Klein, J. C. & Shendure, J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature 518, 120–123.
The mouse embryo was used to evaluate the presence or absence of genetic defects in the human body. Nat. Med. 14, 875–881 (2008).
Huang, C., Li, G., Wu, J., Liang, J. & Wang, X. Identification of pathogenic variants in cancer genes using base editing screens with editing efficiency correction. The full journal of the genetics, genome b.o., 80 (2021).
Calibration of Computational tools for missense variant pathogenogenity classification. Countreads and bioRxiv analysis of BRCA2 variants associated with Fanconi anemia
Mishra, A. P. et al. It’s not possible for RAD51 recruitment at double strand breaks with the BRCA2–DSS1 interaction. Nat. Commun. 13, 1751 (2022).
Pejaver and his wife, V. There are methods for Calibration of Computational tools for missense variant pathogenicity classification. Am. J. Hum. Genet. 114, 1143–1178, 116, 1143–1178
Spurdle, A. B. et al. Towards controlled terminology for reporting germline cancer susceptibility variants: an ENIGMA report. J. Med. Genet. 56, 347–353.
Biswas, K. et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood 118, 2430–2442 (2011).
Arnaudi, M. et al. The assessment is of the structure for the different types ofproteins. Preprints at bioRxiv were published at a rate of 200 per year.
FASTQ files of sequenced samples from Illumina MiSeq or NextSeq assays were trimmed for adapter sequences using cutadapt (v.3.5). The pairs of ends were converted into single reads. The single reads were read and aligned to the human reference genome. The tool Countreads was used for DNA-sequencing data analyses with a focus on identification and characterization. CountReads included the preparation of reference amino acid and DNA sequences, validation of sequencing data integrity and precise trimming of reads to relevant regions. The method also differentiated between variant types and confirmed the presence of specific variants and aggregated and reported variant data. CountReads produced a variant call format (VCF) file, which was annotated using CAVA35. The Spliceai tool was used to evaluate the effects of all the observed SNVs.
The log2 ratio between the frequency of D14 and D0 read counts was used to measure the depletion or enrichment effect for each variant. The comparison of D0 and D5 was used to adjust the position. The read counts that were excluded from further analysis were at D0 and D5. log2 ratios of variants were linearly scaled within each exon across replicate experiments relative to median silent and median nonsense SNV values. The average score was calculated using all of the non-missing values. Linear scaling is similar to within exon normalization in that it uses median synonymous and nonsense values. After completion of all data cleaning and quality control, a raw functional score was available for 6,959 SNVs (Supplementary Table 3).
The P Strong missense alterations were mapped using the PyMol software. The Protein Data Bank source file (identifier 1MJE) was downloaded from the NCBI Molecular Modeling Database. The crystal structure of the complex was used for the three-dimensional modelling.
BRCA2 amino-acid sequences were obtained from Align-GVGD (http://agvgd.hci.utah.edu/). Sequence alignments were performed on ten species, with the exception of Pan troglodytes and Macaca mulatta. The analyses were performed on the parts of the alphabet with the functionally pathogenic variant. They used Align-GVGD26, AlphaMissense 27 and Bayes-Del40 for in silico prediction.
SGE functional results were compared with those from other studies, including a BRCA2-deficient cell-based HDR assay7, a BRCA2-deficient cell line–based drug assay24, a prime-editing-based SGE study16 and a mouse embryonic-stem-cell-based functional analysis25.
The ACMG–AMP rule based framework combines evidence from population, Computational and Predictive, segregation, functional, and other data with each contributing source weighted as very strong. The data gives variant classifications for benign, LB, pathogenic,LP and VUS9. In this study, the ACMG–AMP scoring rules were used in order to determine the clinical classification of the BRCA2 SNVs. The BRCA2 functional data were integrated into the ClinGen–ACMG–AMP BRCA1/2 VCEP classification model under the PS3/BS3 rule. The log scale cap the value for functional evidence to +4 and -4) to make it more difficult for them to be classified as LB orLP. The study was supported by the Western Institutional Review Board, and was not reviewed by the clinical testing cohort. The ACMG criteria used in this study are provided in the supplementary methods.
LOH status for breast, ovarian, pancreatic, and prostate cancer tumours carrying germline BRCA2 DBD variants was acquired from tumour–normal paired sequencing using the IMPACT dataset32. The FACETS algorithm used to determine LOH was used. The analysis only looked at samples with more than 40% tumour content.
Design and characterization of multiple sgRNA targets for SGE based on HAP1 cells and BamHI-HB-digged pUC19
All data shown in this paper are provided with the explicit written consent of the study participants following approval from the institutional review boards.
HAP1 cells were maintained with penicillin and Streppomycin. For haploidy sorting, 1 × 10−7 In order to resuspended the cells, they had to be sorted into various groups at 4 C. HAP1 cells were transfected using Turbofectin 8.0 (Origene). Integrated DNA Technologies made all the primer and oligonucleotides.
The intronic regions flanking exons 15–26 were selected for SGE. Exons 18 and 25 were split into amino-terminal-targeted and carboxy-terminal-targeted regions because of their large exon size, which resulted in a total of 14 SGE target regions. Multiple sgRNAs were designed using the Benchling design tool. The sgRNA-anneals were ligated to p SpCas9(BB)-2A-Puro. For each SGE, 600−1,000 bp homologous arms upstream and downstream of the target region were amplified from wild-type HAP1 gDNA and cloned into a BamHI-HF-digested pUC19 vector using a NEBuilder HiFi DNA assembly Cloning kit. Cloned plasmid backbones were subjected to site-saturation mutagenesis by inverse PCR34 using mutagenized codon NNN primers for all possible nucleotide changes at each amino-acid position. The protospacer adjacent site or the sgRNA recognition site of each target region was introduced by site- directing mutagenesis to prevent re-cutting after successful editing. The introns of each arm were introduced with a single 3-nucleotide fix to facilitate the reamplification of the targeted DNA.