A multi-omic atlas of human development
Using the CITE-Seq Dataset to Analyse and Classify Teratogenic Drugs in Humanoid Cells
On the denoised ADT modalities, annotations of the CITE-seq data was carried out. The scaled highly variable genes were identified using the standard functions in the Seurat package, followed by PCA on the top 2,000 highly variable genes. MNN correction was applied to the PCA loadings matrix using the reducedMNN function from the Batchelor package60 (v.1.18.1) to reduce batch and donor effects between samples. To integrate both modalities, multimodal neighbours and modality weights were identified and a weighted nearest neighbours (WNN) graph58 was constructed using FindMultimodalNeighbors based on the PCA. The number of PCs had to be determined by the variation of consecutive PCs being higher than 0.1%. The weighted combination of both techniques is represented by a UMAP visualization. The sPCA was performed on transcriptome data with the help of the Seurat package to obtain dimensionality reduction for the miRNA that serves the WNN graph.
Drug and target gene information for humans (Homo sapiens) were gathered from the ChEMBL database (https://www.ebi.ac.uk/chembl/). For the teratogenic drugs targeting, we searched the clinically approved molecules that target genes encoding their reported targets and curated a list of 65 clinically approved drugs from the chEMBL database, which carried warnings of teratogenicity (Supplementary Table 6). Drug scores were calculated as previously described58. We came up with a drug category according to the clinical utility. The Drug2cell Python package is available for download.
The method of analysis used in CellphoneDB was called the ‘Cpdb_analysis_method.call’. We included genes expressed in more than 10% of cells within each cluster. Inferred interactions with a P > 0.001 were removed. We used NicheNet (v1.1.1) to identify different interactions between endochondral and intramembranous niches. The Wilcoxon test was used to calculate the DEGs for the clusters of osteogenic tip cells and minimum log fold change was used to summarize their differential expression. The top 1,000 DEGs were used to calculate ligand activities. The default settings were used to prioritize the ligand–receptor links. The top ten ligands and their top-scoring receptors were visualized using heatmaps.
We used a network propagation98 approach to integrate GWAS summary statistics and cell cluster marker gene-based scores for prioritizing disease-relevant and cell cluster-specific subunits of our transcription factor network. First, scores per SNP were computed from downloaded summary statistics and weighted by linkage disequilibrium. SCENIC+ computed the scores using an enhancer-driven GRN which was mapped to the GRN of the corresponding lineage. The networks contain transcription factors and target genes, while regions with transcription factor-binding sites were included in the map of the scores. Using a restart process or random walk, the scores were eventually spread across the network. This integrates the contribution of individual SNPs, with signals converging around relevant network nodes. The procedure was repeated with a further 1000 randomly permuted scores. The cell clusters specificity scores were created by the same process of propagation of differential expression-based marker genes. The scores for the genes were combined per cell cluster. 99) by using the minimum for each node. The connected components were obtained as enriched subnetworks after the final scores were thresholded. The method has been compiled into a tool that we called SNP2Cell, which is available at https://github.com/Teichlab/snp2cell.
fGWAS analysis97 was applied to identify disease-relevant cell clusters as described in detail55 (https://github.com/natsuhiko/PHM). The model uses full summary statistics from GWAS to show a general trend between the expression of genes and their association with diseases. The model corrects for linkage disequilibrium and other factors. We used full GWAS summary statistics obtained from the EBI GWAS Catalog, open targets, and knee and hip osteoarthritis as well as total knee and hip replacement from ref. Supplementary Table 8 can be found at the https://msk. Hugeamp.org website.
Regulons predicted with SCENIC+83 and scFates 85 were imported into CellOracle as base GRNs after osteogenesis trajectory was created with scFates 85. Cells were grouped into meta-cells of 10–15 cells and linear models were used to explain transcription factor from target gene expression. Regulon-based transcription factor perturbation vectors were inferred using the cell cluster-specific models to predict effects of transcription factor overexpression and knockout. Diffusion pseudotime96 was then computed for intramembranous and endochondral ossification lineages separately by selecting corresponding starting points. The pseudotime perturbation cross-product was computed and used to derive a set of differential equations. These perturbation scores indicate whether the in silico perturbation of a transcription factor is consistent with or opposes differentiation along a lineage (osteogenesis). The simulations were carried out with care and precision, over expressing and knocking out all the transcription factors. The scores were averaged per cell cluster and then summarized in a table to screen for transcription factors that promote or impede the growth of the body’s cells.
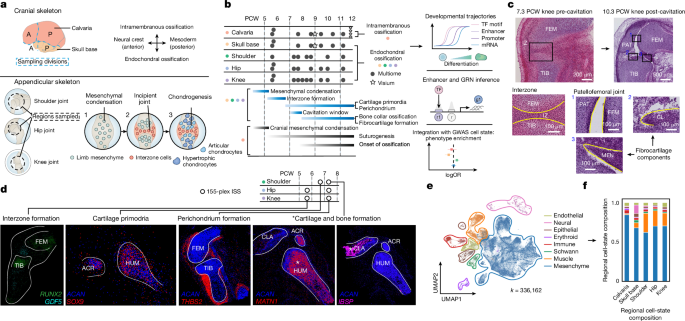
To approximate the timing of cavitation onset, we computed a cavitation enrichment score using sc.tl.score_genes() in scanpy on a specific gene set within the mesenchymal and muscle compartments of the hip, shoulder and knee joints comprising CD44, HAS2, ABCC5, HMMR, MSN and UDPGD, derived from literature and Gene Ontology terms, which encompass hyaluronan biosynthetic processes and hyaluronan synthase activity. The genes that were excluded were with low expression levels. We looked for genes that fit Gene Ontology terms and calculated the correlation between their enrichment scores and cavitation enrichment scores.
OrganAxis in Visium and IBEX: Maps, annotations and approximation using ISS-Patcher
OrganAxis is a mathematical model aimed to derive the relative, signed position of a point in space in respect to two morphological landmarks. The OrganAxis base function H is very flexible and tuneable when it comes to research questions, spatial resolution and sampling frequencies. We give a detailed guide on tissue annotations in Supplementary Notes 1 and 2. The CMA is an extrapolation of OrganAxis, which is defined by a weighted linear combination of two H functions (CMA = 0.2 × H(edge-to-cortex) + 0.8 × H(cortex-to-medulla)). All Visium and IBEX images were annotated with TissueTag v.0.1.1 at a resolution of 2 um perpixel. annotations were transferred to a quasi-hexagonal grid which was generated by placing points with r-micron spacing in the x and y directions and staggering every other row by r/2 The study began with r quotient of 15. L2 distances to broad level annotations (annotation_level_0) and the corresponding CMA values were calculated with k = 10 for all spots in the hexagonal grid. By nearest-neighbour mapping, the distances to annotations were calculated and transferred to spots in Visium or IBEX.
ISS-Patcher is a package for approximating features not experimentally captured in low-dimensional data based on related high-dimensional data. Matching snRNA-sqa data as a reference in this study was used to develop an approach to approximate expression signatures for genes that are not included in the data. First, a shared feature space between both datasets was identified by subsetting the 155–158 genes present in the ISS pool, followed by separate normalization to median total cell counts, log-transformation and z-scoring for both modalities. Then, the 15 nearest neighbours in scRNA-seq space were identified for each ISS cell with the Annoy Python package, and the genes absent from ISS were imputed as the average raw counts of the scRNA-seq neighbours.
Visium data was sieved based on read coverage and predicted cell abundance after deconvolution. There were spots with less than 1,000 genes per spot or fewer than 25 predicted cells. Furthermore, annotated tissue artefacts and areas not assigned to a specific structure were removed. Next, to generate a common embedding we performed scVI integration after removing cell cycle, mitochondrial and TCR genes from the highly variable gene selection. sc VI training was performed with sample id as the key and categorical covariates and age and width as continuous co variables.
A differential abundance test was created using the Python implementation of the MILO package. We used the scVI latent representation to create a k-nearest neighbour graph of droplets in the relevant compartment and subsequently applied milopy to allocate droplets to overlapping neighbourhoods, with these droplets originating from multiple samples (brc_code). Each neighbourhood was then categorized as a group with majority voting. We binarized values for anterior–posterior positions and calvarium-appendicular covariates to allow testing across these variables. We then determined log fold-change values for differential abundance and false discovery rate values based on the Bejanmini–Hochberg correction.
The scFates package 48 was used for trajectory analysis on the combined m2 and m3 cells. mTECII and mTECIII cells from the paediatric scRNA-seq dataset were reintegrated and batch-corrected using scVI. The scVI embedded was input to the program called ‘Palantir.Utils.run_diffusion_maps’. Tree learning was performed using scf.tl.tree by using multiscale diffusion space from Palantir as recommended by scFates. The expression of mTECI markers together with the mTECII markers were used as a root to compute pseudotime. Finally, milestones obtained after running the pseudotime were used to adjust the annotations. To derive differentially expressed genes across pseudotime branches, we applied the scf.tl.test_fork function, followed by scf.tl.branch-specific as described in the scFates article48. In brief, it fits a generalized additive model for each gene using pseudotime, branch and interaction between pseudotime and branch as covariates; two-sided P values were extracted for the interaction term (pseudotime:branch) and corrected using FDR to obtain significant differentially expressed genes. The genes were tested for upregulation in each branch and assigned to different branches based on a 1.3-fold upregulation cut-off.
The fresh frozen sections were stained using the siRNA kit and the LS Multiplex kit by using the automated version of the BOND, according to the statement. All of the sections were subjected to 15 min of protease III treatment before staining protocols were performed. Before running RNAscope probe panels, the RNA quality of fresh frozen samples was assessed using multiplex positive (RNAscope LS 2.5 4-plex Positive Control Probe, ACD Bio-Techne, 321808) and negative (RNAscope 4-plex LS Multiplex Negative Control Probe, ACD Bio-Techne, 321838) controls.
Visium CytAssist Spatial Gene Expression for Fresh Frozen was done following the manufacturer’s protocol. The locations of interest were chosen based on the presence of the microenvironments of bone formation relevant to the droplet data. The Visium CytAssist protocol for subsequent alignment was performed using the images from the Hamamatsu S60 slide scanner. SpaceRanger mapped the libraries with 10X Genomics.
We used the ISS decoding pipeline outlined in Li et al.65. This pipeline consists of five distinct steps. We used the Acapella scripts supplied by Perkin Elmer to create two-dimensional maximum intensity projections for all channels. The first thing we did was register all the cycles using the default parameters. We used a method called CellPose67 (v 3.0) for the cell segmenting. The expected cell size is set to 70 pixels in diameter and further expanded 10 pixels to mimic the cytoplasm. To decode the RNA molecules, we used the PoSTcode algorithm68 (v1.0) with the following parameters: rna_spot_size = 5, prob_threshold = 0.6, trackpy_percentile = 90 and trackpy_separation = 2. Furthermore, we assigned the decoded RNA molecules to segmented cells using STRtree (v2.0.6) and subsequently generated AnnData objects following the approach described by Virshup et al.69. Finally, only cells with more than four RNA molecules were kept for downstream analysis.
An automated RNA slice from the thymus using whole-cell dissociation: cell isolation, cytochemistry and isolation for cell-based diagnostics
We applied whole-cell dissociation to the new donor tissue. Before cell extraction, the sample tissues (approximately 9 PCW shoulder joints) were dissected to obtain bone samples, and soft tissues were microdissected away. The resultant cell suspension was stained with DAPI (Invitrogen) for live-viability, FGFR3 antibody (1:50; MA5-38521, Thermo Fisher Scientific) and TACR3 antibody (1:50; BS-0166R, Thermo Fisher Scientific), and secondary antibodies. Singlet Cells that were DAPI- positive were gated using a BigFoot Cell Sorter and its proprietary software. Double-positive cells were identified with sequential gating for FGFR3 and TACR3. Positive controls for FGFR3 and TACR3 were conducted using mononuclear cells and unstained cells.
For the analysis of the RNAscope, the tissue from the thymus region was taken and separated for Visium sectioning. Sections were cut from the fresh frozen OCT-embedded (OCT, Leica) samples at a thickness of 10 μm using a cryostat (Leica, CM3050S) and placed onto SuperFrost Plus slides (VWR). Sections were stored at −80 °C until staining. The sections were removed from the −80 °C storage and submerged in chilled (4 °C) 4% PFA for 15 min, then acclimatized to room temperature 4% PFA over 120 min. The sections were then briefly washed in 1× PBS to remove any remaining OCT. The sections were air-dried and dehydrated in a series of different percentages, before being used for an automated RNAscope.
Imaris study with the Blaze light-sheet microscope, iDISCO+ protocol and LightSpeed Mode on a mobile laser microscope (Miltenyi Biotec)
The samples64 were cleared using the iDISCO+ protocol. Samples were placed in TPP (Techno Plastic Products) tubes, dehydrated for 1 h in methanol (20%, 40%, 60%, 80% and 100% (2x)) under rotating agitation (SB3, Stuart). The sample volume was approximately five times that of the Methanol volumes. The samples were next incubated in a solution of 67% DCM and 33% MeOH overnight followed by 100% DCM for 30 min at room temperature on a rotator, then put in 100% DBE. Cleared samples were imaged with a Blaze light-sheet microscope (Miltenyi Biotec) equipped with sCMOS camera 5.5MP (2,560 × 2,160 pixels) controlled by Imspector Pro 7.5.3 acquisition software (Miltenyi Biotec). Four different wavelength lasers generated the light sheet, which was 4 m in thickness. Different magnifications of 0.6, 1 and 1.66 were used for the objectives. Samples were supported by a sample holder from Miltenyi and placed in a tank filled with DBE and illuminated by the laser light sheet from one or both sides depending on the size of the samples. LightSpeed Mode was used to acquire these images in a reasonable time and at a suitable resolution. Mosaics of 3D image tiles were assembled with an overlap of 10% between the tiles. The images were acquired in a 16 bits TIFF format. An automatic alignment of tiles was performed when the images were initially processed. Images from stack were converted to a file. To isolate a specific structure in Imaris, we used the surface tool with manual selection, and then used the surface to mask the image. Images and videos were taken by using either the function Snapshot and Animation in Imaris. The areas were coloured with Adobe Photoshop.
Specimens were denatured during 1 week in a solution of 0.500 M of EDTA. The solution was renewed halfway through the incubation period. The samples were washed twice in one day. For one hour at room temperature, the samples were dehydrated in ascending concentrations of methanol in H2O. Then, samples were placed overnight under white light (11 W and 3,000 K°) and rolling agitation (004011000, IKA) with a 6% hydrogen peroxide solution in 100% methanol. Samples were rehydrated for 1 h at room temperature in descending concentrations of methanol (80%, 60%, 40% and 20%), washed twice and blocked in 0.2% PBS-gelatin with 0.5% Triton X-100 (PBSGT) solution during 1 week. A solution containing the primary antibodies was put in a container for 14 days to be cultured at 37 C. This was followed by six washes of 1 h in PBSGT at room temperature. Secondary antibodies were moved through a 0.22m filter. During 1 h in PBSGT the samples were washed six times at a room temperature.
ISS was performed using the 10X Genomics CARTANA HS Library Preparation Kit (1110-02, following protocol D025) and the In Situ Sequencing Kit (3110-02, following protocol D100), which comprise a commercialized version of HybRISS63. The design of the probe panel used folds in the cell state of the limbs. In brief, cryosections of developing limbs were fixed in 3.7% formaldehyde (252549, Merck) in PBS for 30 min and washed twice in PBS for 1 min each before permeabilization. The sections were washed twice in PBS after being briefly digested with a small amount of pepsin. After dehydration in 70% and 100% ethanol for 2 min each, a 9mmdiameter (50 l volume) SecureSeal hybridization chamber was used to hold the reaction mixture on each slide. When rehydration occurred in buffer WB3 the probe hybridization in buffer RM1 took place for 16 h. The probe panels had 5 padlock probes per gene, which are proprietary by 10X Genomics. The section was washed with PBS-T (PBS with 0.05% Tween-20) twice, then with buffer WB4 for 30 min at 37 °C, and three times again with PBS-T. Probe ligation in RM2 was conducted for 2 h at 37 °C, and the section was washed three times with PBS-T, then rolling circle amplification in RM3 was conducted for 18 h at 30 °C. Following PBS-T washes, all rolling circle products (RCPs) were hybridized with LM (Cy5-labelling mix with DAPI) for 30 min at room temperature, the section was washed with PBS-T and dehydrated with 70% and 100% ethanol. The hybridization chamber was removed and the slide mounted with SlowFade Gold Antifade Mountant (S36937, Thermo). Imaging of Cy5-labelled RCPs at this stage acted as a quality control step to confirm RCP (‘anchor’) generation and served to identify spots during decoding. The perkin ederly opera is a high-content screening system that has a 1- m Z-step size. The channels are: DAPI (excitation of 375 nm and emission of 435–480 nm), Atto 425 (excitation of 423 ytd and emission 499 ytd), Cy3 (excitation of 476 ytd and emission 509 ytd The slides were put in PBS with minimal movements to avoid damage to the tissue. The section was dehydrated with 70% and 100% ethanol, and a new hybridization chamber was secured to the slide. The last cycle was stripped with 100% formamide which was washed with PBS-T after 5 minutes. The section was dehydrated, the chamber was removed and the slide mounted. This was done five times to generate the full dataset of seven cycles.
Developing human limb and cranium tissue samples were obtained from elective terminations under REC 96/085 with written and informed consent obtained from all sample donors (East of England, with full approval from the Cambridge Central Research Ethics Committee). During dissection, samples were kept on ice andPBS was used to suspend them. The joints were removed from the limbs. The clavicle and neck of the humerus were the places where a small incision was made for the shoulder joint. When bone features in the glenohumeral and acromioclavicular joints had not formed, approximations were made to capture their entirety. Two regions were separated for each of the calvaria and skull base in the cranium samples, which were less than 8 PCW. For older cranial samples (more than 8 PCW), tissues were dissected to separate the frontal, parietal, sphenoid, ethmoid, occipital and temporal bones where feasible. The samples were initially embedded in the cutting temperature medium and then frozen on isopentane-dry ice. The sections were cut at a thickness of 10 m and placed onto the SuperFrost Plus slides, or directly for single-nucleus processing. Whole-mount immunostaining samples were obtained from aborted pregnancies with written and informed consent of all sample donors. Samples were provided by INSERM’s HuDeCA Biobank and utilized in compliance with French regulations. Full authorization to use these tissues was granted by the French agency for biomedical research (Agence de la Biomédecine, Saint-Denis La Plaine, France; PFS19-012) and the INSERM Ethics Committee (IRB00003888).
Trajectory Inference for T Lineage Cells from CITE-Seq and a New WNN Ultramembrane Analysis
To carry out trajectory inference for αβT lineage cells, CITE-seq data were subsetted to contain DP_pos_sel, DP_4hi8lo, SP_CD4_immature, SP_CD4_semi-mature, SP_CD4_mature, SP_CD8_immature, SP_CD8_semi-mature and SP_CD8_mature cells. A new WNN UMAP was constructed based on surface protein and RNA. Slingshot62 (v.2.6.0) was used to establish a minimum spanning tree on the WNN UMAP using the getLineages function based on mutual nearest neighbour-based distance with DP_pos_sel set as start point and SP_CD4_mature and SP_CD8_mature specified as end points. The derived pseudo time orderings were used for the assessment of transcript and surface marker expression throughout differentiation.
Paired TCR-seq data were processed with Dandelion47 v.0.3.1 and information about productive or non-productive TRA and TRB rearrangements was extracted for each cell to validate cell type annotations after clustering.
The data was processed with the R packages Seurat58, SeuratObject, SeuratDisk, SingleCellExperiment, and Matrix. Data were visualized using ggplot2 (v.3.5.0), ggrastr (v.1.0.2), ggridges (v.0.5.6) and RColorBrewer (v.1.1-3).
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
Cellular cell thawed, pelleted and re-suspended in a magnetic bead-based dead cell removal kit
Cells were thawed slowly by gradually adding 15 volumes of pre-warmed IMDM medium and pelleted at 1,700 rpm for 6 min at 4 °C. After resuspending in PBS, cells were passed through a 70 μm strainer to remove clumps. Enrichment for viable cells was achieved using a magnetic bead-based dead cell removal kit (Miltenyi, 130-090-101). For this, cells were pelleted as before, washed with 1× binding buffer (part of the kit, prepared with sterile distilled water) and resuspended in dead cell removal microbeads (part of the kit) at a concentration of 107 cells per 100 µl beads. After incubation at room temperature for 15 min, cells were applied to an LS column (Miltenyi, 130-122-729), which was prerinsed with 3 ml 1× binding buffer. The column was washed four times and had the flow-through containing viable cells collected. Cells in the flow-through were pelleted and viability was confirmed using trypan blue. 2 106 viable cells were used for TotalSeq-C and Anti-CD3-PE. For this purpose, cells were washed with cell staining buffer (BioLegend, 420201), pelleted at 600g for 10 min at 4 °C and resuspended in 90 µl cell staining buffer. The 10 l Human TruStain FcX blocking solution was added and the cells were prepared for 10 minutes at a temperature of 4 C. After it was resuspended, a supernatant of 25 l was added to the blocked cells. Individual TotalSeq-C antibodies were prepared as described above and 26 µl of the master mix was added to each sample. The samples were added with 10 l anti-CD3-PE to facilitate enrichment and the total staining volume was 200 l. The samples were incubated for 30 min at 4 °C in the dark. The cells were pelleted at 600g for 10 minutes and the staining buffer was added to the samples. All the cells were re-suspended and pelleted as they were before, after removing supernatant. Cells were resuspended in the cell staining buffer and pelleted once more before beingDumpsterDumpsterDumpsterDumpster in 200 l macS buffer In preparation for sorting. Then, 1 µl propidium iodide (Invitrogen, 230111) was added for detection of dead cells and samples were sorted on the BD FACSAria III or BD FACSAria Fusion cell sorter using a 100 μm nozzle and a maximum flow rate of 4 (FACSDiva v.8.0.2, reanalysis with FlowJo v.10.8.2). Cells were gated to remove doublets and debris, then the dead cells were excluded based on their PI staining. The cells were collected separately and cooled, according to the supplementary figs. After completion of the sort, collection tubes were topped up with DPBS and cells were pelleted at 400g for 10 min at 4 °C. The supernatant was removed and cells were resuspended at an estimated concentration of 1,500 cells per µl in PBS + 0.04% BSA (Miltenyi, 130-091-376), of which 16.5 µl was used in the GEM generation step. The next single cell is the gem. Kit v2 (10x Genomics, 1000265) was used to prepare the reaction master mix, and load cells, gel beads and partitioning oil on a Chip K (10x Genomics, 1000286) according to the manufacturer’s protocol CG000330 Rev A. GEMs were generated using a Chromium Controller (10x Genomics), transferred to a tube strip and reverse transcription was performed in a BioRad C1000 Touch Thermal Cycler according to the protocol. The samples were stored at 4 °C overnight and the library preparation was carried out the next day.
Paediatric thymus samples from children undergoing cardiac surgery were obtained according to and used with the approval of the Medical Ethical Commission of Ghent University Hospital, Belgium (EC/2019-0826) through the haematopoietic cell biobank (EC-Bio/1-2018). The tussle was cut into small pieces using scalpels, and then it was fed into an injectionMold to produce a single-cell suspension. The reaction was quenched with 10% FBS and the thymocyte suspension was passed through a 70 μm strainer to remove undigested tissue. Cells were frozen in FBS containing 10% DMSO and stored in liquid nitrogen until needed.
The contrast was used for Extended Data Fig. The following had to be said: DLK2 (magenta 150–500), and LY75 (green 200–4000).
There is a 20 water-immersion objective with 9-11Z-stacks with 2 m step which is used for confocal images. There are five channels: DAPI, Atto 42, Otto 425, and Opal 520. Confocal image stacks were stitched as individual z stacks using proprietary Acapella scripts provided by Perkin Elmer, and visualized using OMERO Plus contains Glencoe Software.
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
Mapping IBEX and CITE-seq DNA using the KNN strategy and GEX: A case study of AIRE+ mTECII
We first used the KNN strategy to identify the IBEX data with high accuracy, and then we added the CITE-seq data as a reference. We used the CITE-seq data as a reference to repeat the KNN-based annotation on these cells. This KNN-based reannotation was performed on a hybrid RNA/protein reference, which included protein measurements for the 19 markers assayed in both CITE-seq and IBEX in addition to RNA measurements for the remaining 23 genes as described in Supplementary Table 11. We used the same KNN implementation as described above but with k = 7, while also imputing CD4 and CD8 pseudotime.
The full example of the KNN mapping using IBEX can be found in the GitHub repository and the general code is found in the dedicated GitHub repository.
In a few IBEX samples AIRE staining produced high levels of non specific signal and low signal-to-noise ratio. As a consequence, we identify some predicted AIRE+ mTECII cells that are not in the medulla and have capsular/cortical localization (Fig. 4g). We had cases of individual samples for which a specific antibody did not perform well or was missing (for example, for IBEX_01 the KRT10 antibody was out of stock). The proper scaling of that marker was not affected by the run of the patcher on each sample. Moreover, by selecting cells with a higher proportion of matched KNN cells from the single-cell data (KKNf) these effects are reconciled through the removal of cells with low-confidence mapping. We would like to flag this point for future researchers who want to use our data.
On the basis of majority voting, the most frequent cell annotation in the GEX reference was assigned to the IBEX query cell. The proportion of KNNs that were part of the majority voting was recorded. The IBEX cell was given the label A if the majority of the 30 nearest neighbours were labeled as A, B, and C.
The KNN of the low-dimensional observations (IBEX) in the high-dimensional space (GEX) were identified. The mean of the high-dimensional neighbours were imputed to the counts of absent features.
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
AnnData v.2.1.1: Combination of IBEX single-cell segmentations and 3D single-nucleus tissue imaging
All sample single-cell segmentations were combined to make up more than one million cells in a unified AnnData object.
We derive the mean and maximum signal intensity for each channel from the IBEX image. This produced a cellXprotein file for each sample.
The high nuclear density produced scenarios in which segments were often bordered by other cells, resulting in signal bleed. To overcome this issue, we excluded pixels in the interface between two cells such that the mask boundary was preserved if no neighbouring cell was present.
After restitching tiles, masks were used to store over 65,535 mask labels in a 32Bit image format, which was then used to produce 200,000–700,000 cell masks.
IBEX images were converted from .ims format (Imaris, Oxford Instruments, v.9.5.0 and v.10.0.0) to 3D stacks (TIFF) by individual channels with FIJI v.1.54j. We then applied a custom pipeline for 3D single nuclei segmentation with cellpose v.2.1.1:
Representative sections from different tissues were acquired using a confocal microscope with a 40 objective (NA 1.3), 4 hyD and 1 PMT detectors and a white-light laser that produces a continuous spectral output between 470 and 670 miles per hour. There were panels with antibodies conjugated to the following dyes: Hoechst, alexa Fluor (MF)487, iFluor 594, and eF 670. The images were captured at an 8-bit depth and have a line average of 3 and a resolution of 1300 m with the following dimensions: x, y and Z. Images were tiled using the LAS X Navigator software. For IBEX tissue imaging, multiple tissue sections were examined before selecting a representative tissue section that contained several distinct lobules with multiple functional units, often resulting in an unusually shaped region of interest. Each detector had different types of lasers that could be used to pick up fluorophore emission which was collected on separate detectors. The Channel Dye Separation module was used to correct for residual spillover. For publication-quality images, Gaussian filters, brightness/contrast adjustments and channel masks were applied uniformly to all images. SimpleITK55.56 was used for image alignment of the IBEX panels. Additional details on antibodies, protocols and software can be found on the IBEX Knowledge-Base (https://doi.org/10.5281/zenodo.7693279).
To identify genes exclusively expressed in a specific cell type or subset thereof (‘specialization genes’, SGs), we developed custom Python functions. Starting from raw read count, gene expression was scaled with scipy.stats.zscore(). The cells that were below a cut-off of 0.05 and the genes that were below 1.5 mean counts, were excluded from further steps. A quantile threshold was used to select cells that have the highest expression level of a specific gene. A χ2 test (scipy chi2_contingency) was performed per gene to identify if the selected cells were over-represented in a specific cell type (cell_type_level_4_explore), indicating the gene to be a marker gene. Genes that were predicted to be expressed only in a single cell type (χ2 α = 1 × 10−50) were considered to be SGs and used for further analyses.
To estimate the dependency of the axis on spot gene variance across samples, we first normalized to a target sum of 2,500 counts and performed log transformation followed by combat regression (https://scanpy.readthedocs.io/en/stable/api/generated/scanpy.pp.combat.html) by sample to adjust for the batch effect of individual samples. We then computed the PCA for batch-corrected gene expression and calculated the Spearman correlation between the first ten PCs and CMA or the number of genes detected per spot (n_gene_by_counts). Note that number of genes per spot, in our hands, was mostly influenced by inconsistent permeabilization during Visium library preparation and constituted the largest technical source of within-sample variance that we found in both fetal and paediatric Visium samples. In order to estimate the cumulative contribution, we divided the Spearman’s R with the first 10 PCs’ cumulative explained variance.
To compare the distribution of cytokines across developmental groups (fetal versus paediatric) and identify differentially distributed genes, we implemented a two-way ANOVA approach. We initially log-normalized Visium gene expression, then removed lobules for which not a single cortex or medulla Visium spot was detected to increase CMA confidence in both datasets. Data were grouped by mean expression per sample and CMA bin, such that each sample had a single datapoint per CMA bin; n = 16 (paediatric) and n = 12 (fetal) samples. The conjugate similarity was calculated from the median values of the pooled sample bins between fetal and paediatric genes. Two-way ANOVA was calculated forfetal and Paediatric ages with the help of a model. P values for main and interaction effects were Bonferroni corrected with statsmodels.stats.multitest.multipletests (pvals, alpha=0.05, method=‘bonferroni’). Supplementary Table 5 can be used to read the full report of the results.
Before performing any association analysis with the CMA, we further removed lobules (based on ‘annotations_lobules_0’) that had no or small medullar or cortical regions, as we expected our CMA model to be less accurate in these cases.
TissueTag v. 0.1.1 was used for semiautomated image annotations. Cortical and medullary cells were predicted with a random forest classification that uses an array of annotations based on the highest gene expression in each cell. Automatic cortex/medulla annotations were then adjusted manually where necessary. The structures and individual thymic lobes were annotated with the help of a machine. The evaluation and morphological annotations were done by experts. A full example of the Visium processing pipeline and annotation is provided on the GitHub repository for this Article (see Code availability).
To process the Visium histology image data in higher resolution than the SpaceRanger defaults, we built a custom pipeline to extract an additional layer of image resolution at up to 5,000 pixels (hires5K), which we found to be more suitable for morphological analysis. We developed a fiducial image registration process that can be used to detect fiducials and register them. Otsu thresholding was used for flexible tissue detection.
Source: A spatial human thymus cell atlas mapped to a continuous tissue axis
The differentiation potential of mcTEC progenitor cells and its application to genomic mapping and data processing for scRNA seq and CITE-seq
To provide a common reference, we also binned the axis to a sequential discrete space for the entire thymus by ten levels (capsular, subcapsular, cortical level 1, cortical level 2, cortical level 3, cortical CMJ, medullary CMJ, medullary level 1, medullary level 2, medullary level 3; details are provided in Supplementary Note 2 and Supplementary Table 8).
The STEMNET package50 (v.0.1) was used to determine the differentiation potential of mcTEC progenitor cells. For this purpose, only three cells in the dataset were retained. mTECI/II/III and cTECI/II/III were set as maturation endpoints and the probability of each cell to adopt any of these six possible fates was calculated. The difference in the probabilities between the two fates for each cell is what we used to derive a priming score. Cells with priming score <−0.5 or >0.5 were labelled as mTECI-primed and cTECI-primed, respectively, whereas all other cells with comparable mTECI and cTECI potential were deemed to be unprimed.
data from several previous studies were included and reanalyzed. Information on sample processing, ethics and funding can be found in the publications and in Supplementary Tables 1 and 2.
Metadata about scRNA-seq and CITE-seq samples, including information on source study, cell enrichment and donor age, are provided in Supplementary Table 1. Information about spatial data, including Visium, IBEX, RareCyte and RNAscope, is provided in Supplementary Table 2. The data was mapped to the human genome after it was generated at Ghent University. All original scRNA-seq data and 10x Visium data were mapped to the human genome at thewsi. All IBEX imaging was performed at the National Institute of Allergy and Infectious Diseases (NIAID), NIH. All non-IBEX imaging datasets (RareCyte, RNAscope, Visium H&E) were generated at WSI. Fetal work was not done at the two universities.