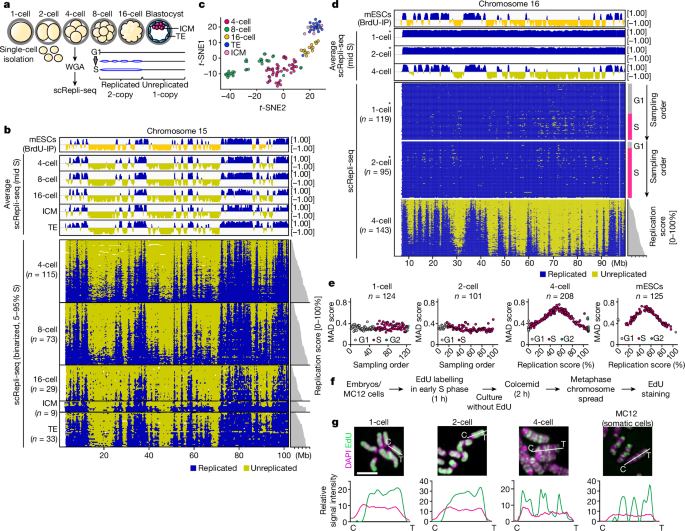
There was embryoploidy instability when DNA replication timing program emergence
Statistical Analysis of Blind and Ad-hoc In-vivo Experiments in a Random Environment Using GraphPad Prism and Analysis of Sample Sizes and Variances
The experiments were duplicated at least twice with the same conclusion, and all of the in-vivo experiments were duplicated in two separate groups. The exact numbers of replicated experiments are stated in each figure legend. Random allocation of samples and animals to experimental groups resulted in research data being collected and analysed blind or ad hoc.
GraphPad Prism v.9.5.0 was used for statistical analysis. Mean and s. e.m. were used to represent the sample. Chi-squared and paired two-tailed Student’s t-tests were used to compare two samples; analysis of variance was used to compare multiple data points. No statistical methods were used to predetermine sample sizes.
Quantification of adipocyte size in WAT and iBAT with Fiji script using Analyse Particles and Otsu thresholding
The size of adipocytes in WAT and iBAT was quantified as previously described55 using Fiji. WAT was stained with haematylin and eosin and then scanned into a digital image using a zoomser-sq Digital slide scanner. Images were randomly picked and the plug-in Analyse Particles was used to count the number and measure the size of adipocytes and droplets in iBAT. 11,000 cholesterol droplets were measured for each iBAT.
The percentages of PDGFRα+ and DES+ cells were counted manually based on PDGFRα and DES signals. There was a count of the percentages of NPY+ and Th+ neurons in the hypothalamus. To calculate ganglionic mural cell coverage, 531, 531 and 30 μm xyz views were randomly picked and maximally projected to z using Fiji script. Labelled areas were segmented using a threshold set by default, and coverage was calculated as area NPY+/area CD31+.
The percentage of EdU+) cells was calculated with an automatic method. The thresholding method was used to set it.
The tool Imaris used to measure the innervation of NPY+ axons was called the Surface. Labelled areas of NPY+ axons and CD31+ vessels in whole cleared iWAT were automatically segmented, and the innervation of NPY in vasculature was calculated as volumeNPY+/volumeCD31+. The total coverage was calculated as volumeDES+/volumeCD31+. The images showing NPY+ innervation were quantified with an automatic unbiased method. The Otsu thresholding method was used to segment areaNPY+ and areaCD31+.
The overlapping between NPY and CD31 was calculated using a plug in. To make this calculation, 472.33, 472.33 and 30 μm xyz regions were randomly picked and maximally projected to z using Fiji script. A threshold was set by default and the percentages were calculated automatically.
Source: Sympathetic neuropeptide Y protects from obesity by sustaining thermogenic fat
Isolation and growth of mural cell-specific medium (Science Cell) based on public scRNA-seq datasets
Public scRNA-seq datasets were downloaded from Gene Expression Omnibus (GEO) and analysed using the following method. The cells with less than 200 unique detected genes were discarded. The normalized cell matrix was created using the Seurat v.4.2.0. 50) in R (v.4.2.2). Data were then scaled using ‘ScaleData()’, and linear dimensional reduction performed by principal component analysis and calculation of UMAP coordinates for all cells using Seurat v.4.2.0. Cells were clustered using ‘FindNeighbour()’, with dimensions set to 15, and ‘FindClusters()’, with resolution set to 0.5. Each cluster was identified based on differentially expressed genes and known markers in the published literature5,22,29,51,52,53.
The cells were isolated and then cultured in a different way. In brief, mice were euthanized intraperitoneally with 10 μl g−1 pentobarbital and perfused with 15 ml of PBS to remove blood. The iBAT SVF was isolated using the method described above. Cells were then placed in 10 cm dishes with 10ml of DMEM and 10% FBS for 2 days to attach. Afterwards, 5 ml of the medium was replaced with mural cell-specific medium (DMEM with 2% FBS, supplied with 1% mural cell growth supplement (Science Cell, catalogue no. 1252)). Every other day, 5 ml of medium in dishes was refreshed until the cells grew confluent. For the purpose of acquisition of mural cells, cells were labelled with an anti- CD104b and sorted with a magnetic buffer.
Cells from 12well plates had a density of 1 105 per liter for 3T3-L1 cells to be differentiated into white adipocytes. The medium was refreshed after the cells confluent. Two days following cell confluence, induction medium (10% FBS, 500 μM 3-isobutyl-methylxanthine and 1 μM dexamethasone in DMEM) with or without 1 μg ml−1 insulin was added to each well; 3 days later the induction medium was replaced with maintenance medium (DMEM with 10% FBS with or without 1 μg ml−1 insulin). Subsequently the medium was refreshed with maintenance medium every 2 days. Total differentiation time for each well was 8 days. NPY treatments started when the medium was added, and lasted throughout the process.
There is no testing for mycoplasma contamination in the mouse macrophage cell line. The complete medium for cell culture contains high-glucoseDDM with glutamate and 10% FBS.
The cells wereseeded at a rate of 5 104 per millimetres on the covers of a glass container to perform the coculturing experiments. 220-BB) and NPY (Cayman Chemical, catalogue no. CAY15071) were added to cells 24 h following cell seeding. Cells were collected 5 days later and either fixed with 4% PFA for imaging or lysed with Trizol for RNA extraction and qPCR.
To remove red blood cells, the cells were treated with a red blood cell lysis buffer and then treated with Fc block. 14-9161-73) before staining with antibodies. The eBio science used a Buffer Set for the permeabilization of cells. Cell sorting was done using the FACSAria III sorter, as well as flow cytometry data was obtained from either a BD LSRFortESSA X20 or any of the above software. The data was analysed using FlowJo.
The mice were euthanized using 10 l g1 pentobarbital and the blood was collected from the left ventricle with a 25G needle and a 100 mM EDTA vaccine. The blood was placed in tubes for 100 mM. EDTA and centrifuged at 1,000× rcf at 4 °C for 15 min to separate plasma from blood cells, and then the supernatant was transferred to fresh tubes with DPP-IV inhibitor (final concentration 50 μM) and aprotinin (final concentration 500,000 IU ml−1). The concentration of NPY was determined using an NPY ELISA kit. The name of the game is EZRMNPY. The data was recorded using a FLUOstar Omega microplate reader.
IWAT’s proteins were derived as was previously described. In brief, dissected iWAT was placed in homogenizing tubes filled with 300 μl 50 mg−1 RIPA lysis buffer (Sigma, catalogue no. 20188) containing 50 μM DPP-IV inhibitor (Sigma, catalogue no. The 500,000 IU aprotinin is found in the catalogue no. A 6103-1MG and a Precellys 24 homogenizer were used. The clear portion of the fat was retained after it was removed by centrifuging at 20,000 rCF for 15 minutes. The process was repeated three times to ensure complete removal of lipid.
RNA extraction was performed using Trizol reagent (ThermoFisher, catalogue no. 15596026), complementary DNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen, catalogue no. 18064022) and qPCR was performed using Power SYBR Green PCR Master Mix (LifeTech, catalogue no. 4368706). There was data recorded using the StepOne system and analysed with Stepone software. Primers used in this research are listed in Supplementary Table 1.
The ΔCt method was used to quantify gene expression using the following formula: relative expression = 2^ (−(Cttarget gene − Ctreference gene)). The ΔΔCt method was used to compare the thermogenic and adipogenic gene expression shown in Fig. 5b and extended data figs are included. 9l,m and 10m are included.
The following primary antibodies were used for immunofluorescent staining: rat anti-CD31 (BioLegend, catalogue no. 102501, MEC13.3, 1:100 dilution), rat anti-PLVAP (BioLegend, catalogue no. 120503, MECA32, 1:100 dilution), rabbit anti-DES (abcam, catalogue no. rabbit anti-NPY, catalogue no. D7Y5A, 1:500 dilution. ab30914, 1:500 dilution), chicken anti-TH (Aves Labs, catalogue no. TYH73787982, 1:500 dilution), rabbit anti-TH (Sigma, catalogue no. Ab152, 1:500 dilution), mouse anti-NPY1R (Santa Cruz, catalogue no. sc-393192, 1:200 dilution, rat anti-PDGFR. ab14106, 1:250 dilution), Cy3 anti-αSMA (Sigma, catalogue no. C6198, 1A4, 1:250 dilution), goat anti-SOX17 (R&D, catalogue no. AF1924, 1:250 dilution), goat anti-EPHB4 (R&D, catalogue no. rabbit anti-UCP1 and AF3034 are included in the catalogue.
Source: Sympathetic neuropeptide Y protects from obesity by sustaining thermogenic fat
Development of epididymal white adipose tissue using dextran and a confocal laser microscopy
Animals were anaesthetized with ketamine (100 mg kg−1) and xylazine (10 mg kg−1) and a small incision was made to expose epididymal white adipose tissue for microscopy analysis. The images were obtained by means of an optical microscope with coherent anti-Stokes Raman scattering, a two-photon excimer and a bright-field microscope. All procedures were performed at the National Institute of Science and Technology on Photonics Applied to Cell Biology at the State University of Campinas. Two lines of lasers with the same wavelength were used to acquire adipocytes images from the anti-Stokes scattering. The T1162 is a book The wavelength for the Stokes is 1,040. After ten frames, 50 l (30 g/d) of the drug was obtained and put into the video after identification of blood vessels. The mouse had a T1162) injected into it. The video kept recording for about 12 minutes. The duration of the video was analysed forescence intensity. The image was analysed after dextran administration. The region of interest inside the vessel was selected for the evaluation of tissue leak. In both areas the intensity was measured and the ratio between the tissue and the vessel was calculated. Blood flow was calculated using the average of the raw intensity of dextran inside the artery over 100 s immediately following stabilization of the dextran signal after administration.
Images were acquired using a Zeiss LSM880 confocal microscope with Zen-black software (v.2.1). A 20/0.8 NA objective was used, and samples were imaged using either a x,y, or z 10 m. The solid-state lasers of 405, 609 and 588 were used for the DAPI.
For staining, whole-mount, PFA-fixed ganglia were dehydrated once in each of 30, 50, 70 and 90% ethanol, twice in 100% ethanol—with shaking for 20 min at each concentration—and then rehydrated in 90, 70, 50 and 30% ethanol. Ganglia were then chewed up with 1.5 U ml 1 dispase-1 and 500 g hyaluronidase. Ethanol-treated ganglia were blocked in blocking solution (3% bovine serum albumin, 2% goat serum, 0.1% Triton X-100 and 0.1% sodium azide in PBS) for 2 h and then incubated with primary antibodies diluted in the permeabilizing buffer for 3 days at 4 °C. Three days later, ganglia were incubated in secondary antibodies diluted in the permeabilizing buffer for a further 3 days at 4 °C. ganglia were dehydrated once each in 30, 50, 70, and 90%. They were cleared using ethyl cinnamate (ECi). Cleared ganglia were mounted in a coverslip slide sandwich filled with ECi.
For paraffin embedding, fixed samples were processed in 70, 80 and 90% ethanol and then in 100% ethanol, 100% xylene and 60 °C paraffin wax, three times for each condition. Samples were embedded in paraffin wax, sectioned at 7 µm, mounted on coverslips and dried in an oven at 45–50 °C overnight. Before staining, samples were deparaffinized twice in 100% xylene for 5 min, rehydrated twice in 100% ethanol and then once each in 95 and 80% ethanol and water for 10 min. Adding 0.05% trypsin to each slide and incubateging at 37 C for 20 min was all it took for the antigen to be retrieved. After washing with running water, samples were stained using the same procedure as for fixed cells.
For cryosection, samples were first embedded in OCT (VWR, catalogue no. 361603E) and frozen at −20 °C until cryosectioning. Samples were cut into 10-µm-thick sections and mounted on charged SuperFrost Plus slides (VWR, catalogue no. staining using the same procedure as for fixed cells
Superior cervical ganglia and sympathetic axon bundles were dissected from adipose tissues of mice perfused with 20 ml of PBS, and the tissues were fixed in 4% paraformaldehyde (PFA, 043368.9 M, ThermoFisher) for at least 4 h. Cells collected following in vitro experiments were washed once with PBS and fixed using 4% PFA for at least 4 h. Axon bundles were first stained by incubation overnight at 4 °C with primary antibodies dissolved in the permeabilizing buffer (3% bovine serum albumin, 2% goat serum, 0.1% Triton X-100 and 0.1% sodium azide in PBS), followed by incubation with secondary antibodies and DAPI dissolved in the permeabilizing buffer for 1 h with gentle agitation. As for immunofluorescence in fixed cells, cells were first incubated with the permeabilizing buffer for 1 h at room temperature and then stained overnight at 4 °C with primary antibodies diluted in the permeabilizing buffer. The samples were then washed and stained for 1 h at room temperature. Sympathetic fibres or fixed cells were then mounted on slides with Fluoromount-G mounting medium (ThermoFisher, catalogue no. 00-4958-02).
For viral injection, mice were anaesthetized with avertin (240 mg kg−1 intraperitoneally) and fixed on a stereotaxic holder (RWD, Life Science). Either AAV2/9-hSyn-Cre-EGFP-WPRE-pA (Taitool, catalogue no. There are 2 1,012 viral genomes or AAV2 (9-hSyn-EGFP- WPRE-pA). S0237-9, Taitool, 2 × 1,012 viral genomes ml−1, 200 nl per side) was bilaterally injected into the nucleus tractus solitarius (NTS) (NTS coordinates anteroposterior, mediolateral, dorsoventral −7.5, ±0.35 and −4.75 mm, respectively) of Npyflox/flox mice.
Source: Sympathetic neuropeptide Y protects from obesity by sustaining thermogenic fat
Thermoimaging of 6-week old animals (B6.Cg-7630403G23RikTg(Th-cre) 1Tmd/J) and Cx3cr1GFP
Animals as young as 12 weeks were used for thermoimaging. The data was recorded using an Optris thermo camera, and analysed with FOTRIC software. Animals were single housed and placed under the thermocameras, with their back shaved to expose the skin above iBAT. Animals were acclimatized for 4 days in individual cages with ad libitum access to food and water at room temperature under a 12/12 h light/dark cycle before a 14-h fast. Mice had ad libitum access to water during fasting.
iBAT temperature was recorded every 1 s over a 6-day period by storing the temperature of the warmest pixel in view using the software provided by the camera manufacturer (rel. 2.0.6, Optris PIX Connect). The average of temperatures during the 1 h period was iBAT temperature at 14 h.
The calorimetry system was used to analyse the oxygen use, carbon dioxide production, energy expenditure and respiratory exchange rate of the 6 week old animals. Animals were maintained in individual cages following a 12/12 h light/dark cycle with water and food supplied ad libitum, and under controlled room temperature (21–23 °C) and 50% humidity. The data was collected for 5 days afteracclimatation. Animals’ body weight (g) and food (g) were measured before entering and after exiting the cage. Data were normalized by weight using the CalR Web-based Analysis Tool for Indirect Calorimetry Experiments.
Both ThCre mice (B6.Cg-7630403G23RikTg(Th-cre) 1Tmd/J) and Cx3cr1GFP/+ mice were found. Npyflox/flox mice were a donation from the I. Kalajzic Laboratory at the Department of Reconstructive Science, University of Connecticut44 under the material transfer agreement (MTA). The tissues of NPY-GFP mice were obtained from the Brandy Memorial Laboratory at Yale University. M. Roberts is a otosychiatrist at the Department of head and neck surgery at the University of Michigan. ThCre mice were crossed with Npyflorx/flox mice to create sympathetic neuron-specific NPY-cKO mice. Diet-induced obesity was achieved by feeding mice an HFD (Diet Research, product no. They had a feeding regime for 10 weeks when they were 7 weeks old. Each cage had a recorder which records the body weight and food consumption of the mice. All mice were group housed in standard housing under controlled room temperature (21–23 °C) and 50% humidity under a 12/12 h light/dark cycle and given access to diet and water ad libitum. The project licence for all experimental procedures was obtained in accordance with the UK ANIMALS ACTS 1986. P80EDA9F7) and personal licences granted by the UK Home Office. The University of Oxford has an ethical review panel.
Probing cell division that produced de novo chromosome errors in embryos by using AneuFinder with 500 kb bins
Experimental reproducibility was demonstrated as follows: Fig. 1g, two independent experiments; Fig. 2d, two (2-cell) and three (1-cell, 4-cell) independent experiments. 2l, two independent experiments; Fig. Two independent experiments are in this picture. Three independent experiments and two independent experiments are shown in the photo. 4h, two independent experiments and Fig. 4i, two independent experiments.
To analyse chromosome aberration, we used the findCNVs command in AneuFinder with a 500 kb bin size as described previously16 (6-HMM options: method=“HMM”, max.iter=3000, states=c(“zero-inflation”, “0-somy”, “1-somy”, “2-somy”, “3-somy”, “4-somy”, “5-somy”, “6-somy”), eps=0.01). Using scRepli-seq data of 4-, 8- and 16-cell-stage embryos, we determined the cell division that produced de novo chromosome errors as follows. A pair of cells within an embryo that exhibit a 3:1 copy-number ratio for a particular chromosomal region are judged as a pair of sister cells that experienced a chromosome segregation error. 3b is a de novo chromosome abnormality. When identical chromosomal abnormalities were commonly found in multiple pairs of cells within an embryo, these cells were judged to have experienced errors in divisions preceding the last division. The details of all the detected chromosomes are in an extended data fig. There are 6b and Supplementary Table 1. Our dataset is not suitable for female-to- female variability assessment because we pooled embryos from multiple females before single embryo sampling.
Principal component 1 (PC1; A/B compartment profile) of Hi-C data in 200 kb bins was computed from the .hic file using published mapped Hi-C datasets of sperm68 and cumulus60 cells as described previously16. The genomic coordinates of PC1 profiles were converted from mm10 to mm9 using the liftover tool (UCSC Genome Browser). In Extended Data Figs. 2k and 4c, we defined the four PC1 (A/B compartment) categories, A1, A2, B2 and B1, as those containing 25% of all PC1 values (200 kb bins) each from the highest (strongest A) to lowest (strongest B) without an overlap.
The timing of each embryo’s fertilization was recorded by stereomicroscopy every 30 min. At each hour after fertilization or cleavage, embryos were collected, treated with 20 µM EdU for 30 min, and then fixed in 3.7% paraformaldehyde in PBS-PVA (pH 7.4) for 30 min. EdU staining was performed using Click-iT Plus EdU Alexa Fluor 555 or 594 Imaging Kit (Invitrogen). After EdU staining, samples were washed multiple times in PBS-PVA-BSA and then incubated with mouse anti-histone H3 at a temperature of 4 C. The embryos were finally washed and transferred to BSA–PVA for imaging on the Zeiss LSM780 confocal microscope. Imaris software was used to create a 3D version of the images. In Fig. There is a 2b–d and extended data fig. 3a–c, we categorized EdU staining patterns based on spatial distribution and intensity. Images of nuclei were subjected to auto-thresholding with the ‘default’ algorism in Fiji software, and the thresholded patterns were manually categorized into ‘uniform’, ‘nuclear periphery + NPBs’, ‘NPBs’, ‘nPBs + internal foci’, ‘nuclear periphery + internal foci’, and ‘internal foci’. The strong and weak groups were split into two groups, with those with nuclear EdU intensity 1.5 higher relative to the cytoplasmic intensity. The EdU intensity was considered a no signal if it was less than the cytoplasmic intensity. The experiment is shown in a data diagram. 3a, EdU-stained cells after Hoechst 33342 treatment (20 μM, for 30 min at 37 °C) were analysed using fluorescence-activated cell sorting using the Sony SH800 cell sorter using the single-cell mode (SH800 v.2.1).
For the experiments described in Extended Data Fig. The cells were cultured in a glass-bottomed dish, which was subjected to 0.5 M nocodazole for 17 h to sync with the prometaphase. After synchronization at the G1/S-phase border, the cells were further cultured in the presence of aphidicolin for 15 h with or without 10 mM 2-aminopurine (2-AP; an intra-S-phase checkpoint inhibitor). After removing aphidicolin or 2-AP, the cells were labelled with 20 μM EdU for 60 min and stained with EdU. The C57BL/6 strain 4-cell and 8-cell embryos within 1–2 h after the 2-to-4-cell or 4-to-8-cell division (that is, in early S phase) were treated with 3 μg ml−1 aphidicolin with or without 2-AP for 5.5 h followed by reagent removal and EdU staining.
Source: Embryonic genome instability upon DNA replication timing program emergence
Auto-thresholding measurements and determination of fork speed of mobile fork class fibres of the DNA and of their cytoplasmic and histone H3 levels
Images of the DNA were subjected to auto-thresholding. The individual DNA fibres were shown to be part of a series of dot signals. The fibres containing the signals were categorized into those with and without class forks. The class forks with only one dot signal of IdU + CSD or CSD with gaps between dots reflect extremely slow fork movement. The gaps were defined as those with at least one dot of the ssDNA signal. I am mobile. The classes with a series of dots were called the mobile class forks. that contain a series of dot signals (≥2 dots) of CldU on either side of the same DNA fibre (Fig. 2h (mobile)). The intermediate class forks were defined as those with an intermediate character between the two other classes; these fibres also contained dot signals of IdU + CldU and CldU with gaps between dots but also contained some IdU + CldU single colour signals (that is, IdU-only regions) in between these dot signals. The measurement method on the mobile fork class fibres is provided. 5b. The calculation of the IOD between the forks was done with the help of the Fiji software. It was possible to determine the fork speed of the mobile forks with the help of the tracks flanked by the IdU tracks. CldU tracks that had no ssDNA signals ahead of them were excluded from the fork speed measurement as these fibres may have been broken in the middle of the CldU track. The details for the immobile fork speed measurement can be found in Supplementary Note 2.
The mean signal intensity was obtained for H3K9me2, p-CHk1 or H2AX, when compared to levels of histone H3. We then subtracted the mean cytoplasmic signal intensity (Ime_cyto), which was obtained from a region near the nuclei, from the Ime_nuc value (Ime_nuc − Ime_cyto). Similarly, we determined the histone H3 level within the same nuclei (IH3_nuc − IH3_cyto). We calculated the ratio between the two values.
Embryos were fixed with 2% paraformaldehyde in PBS-polyvinyl alcohol (PVA) (pH 7.4) for 30 min. The embryo was washed in PBS-PVA-BSA after being incubated with appropriate primary antibodies. DNA was counterstained with 40 µg ml−1 of Hoechst 33342. Finally, the embryos were washed and transferred to BSA-PVA for imaging with a Zeiss LSM780 confocal microscope. There were three primary antibodies used: mouse anti- H2A.X, rabbit anti-histone H3 and mouse anti-histone H3. The secondary antibodies were Alexa Fluor 488 goat anti-mouse IgG (H+L) (A11029); goat anti-rabbit IgG (H+L) (A11034); Alexa Fluor 555 goat anti-mouse IgG (H+L) (A21424) (1:400, Invitrogen).
Experiments using mouse embryos were reported to have slight modifications. In brief, unfixed single blastomeres (derived from BDF1 or B6MSM strain embryo), were collected into 8-wellPCR tubes with 6 l cell lysis buffer. We analysed the blastomeres of the embryo only if there was accidental damage to the cell. For scRepli-seq experiments after EdU staining (Extended Data Fig. After taking the photographs, the cells were put in 12 l of cell lysis buffer and incubated at 55 C for 16 hours. The manufacturer’s instructions were used to complete the remaining whole-genome amplification process and theNGS library construction. The samples were processed for NGS on the Illumina Hiseq 1500 or Hiseq X Ten system (80-bp-length single-read or 150-bp-length paired-end read sequencing).
The mouse cages had a temperature, humidity and light cycle of differing degrees of darkness. Mature oocytes were collected from the oviducts of eight- to ten-week-old female mice that had been induced to superovulate with 5 IU of equine chorionic gonadotropin (eCG, ASKA Pharmaceutical) followed by 5 IU of human chorionic gonadotropin (hCG; ASKA Pharmaceutical) 48 h later. Cumulus-oocyte complexes were collected from the oviducts approximately 16 h after hCG injection. Cumulus-oocyte complexes were placed in M2 medium and treated with 0.1% (w/v) bovine testicular hyaluronidase. After several minutes, the oocytes were washed and then moved to different medium. Mature metaphase II (MII) oocytes were subjected to in vitro fertilization (IVF), intracytoplasmic sperm injection (ICSI) or SCNT. Developmentally arrested or delayed embryos at each embryonic day were excluded from further analysis. The ovaducts from pregnant mice that were stimulated to have sex with male mice were collected for embryo development. In vitro zygotes were cultured in CZB. We pooled multiple females before single-embryo sampling so the dataset is not suitable for assessment of female to female variability. When indicated, embryo were cultured in the presence of nucleosides (Sigma-Aldrich), at a final concentration of 2 or aphidicolin. In Extended Data Fig. embryos were monitored under a microscope every 30 minutes, and if they were deemed to be in G1, they would be transferred to the medium containing aphidticolin. Live images were tracked by the M phase in the extended data fig. 7a. The 4-to-8-cell division of the embryo was just completed when it was collected for scRepli-seq. 7b.
The guidelines for the care and use of animals in laboratory animal experiments were approved by the Institutional Committee of laboratory animal experiments. The production of oocytes and sperm was accomplished by using B6D2F1 and C57BL/6 mice. For allele-specific analysis, C57BL/6 female mice and MSM/Ms male mice were used to produce oocytes and sperm, respectively. To eliminate the effect of individual differences between mice, multiple mice were used in each experiment as follows. There are 2, 16 and 8 ICM andTE mice in Figure 1b. 10 mice each, 1d, 2d, and 4d. 3, 2, and 4 mice, and Fig. 2f, 3, 1, and 4 mice. 3b, 4 in vitro and 2 in vivo (2-to-4-cell), 8 in vitro and 7 in vivo (4-to-8-cell), and 4 in vitro and 2 in vivo (8-to-16-cell) mice; Fig. 3c, 15 mice each (2-to-4-cell, 4-to-8-cell and 8-to-16-cell); Fig. 3g, 3 mice each (2-to-4-cell, 4-to-8-cell, 8-to-16-cell and 16-to-32-cell); Fig. 3h, 4 (2-cell) and 3 (4-cell and 8-cell) mice; Fig. 3i, 5 (2-cell, 4-cell, and 8-cell) mice; Fig. 4d, 4 mice; Fig. 4h–i, 6 mice; Fig. 4j, 16 mice; Extended Data Fig. 7a, 7 (2-to-4-cell DMSO), 6 (2-to-4-cell aphidicolin 15 ng, 30 ng, and 60 ng, and 8 2-to-4-cell aphidicolin 75 ng)

