Evaluating the evidence for adaptive rate variation
Errors and biases in somatic mutation accumulation in genes, genes and essential genes: a meta-analysis of germline mutations
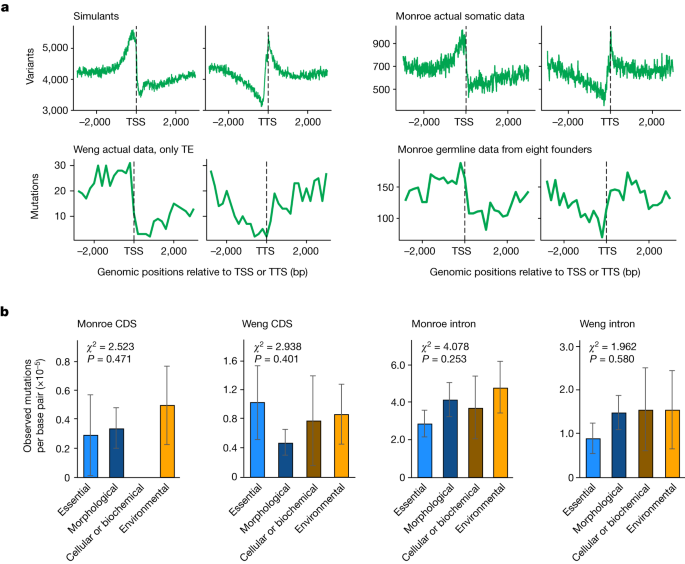
We do not suppose these to be all of the errors. Whereas Monroe call 773,141 mutations using our sequence11, using the same HaplotypeCaller-GVCF calling method12, with default parameters and without any further filtering, we identify only 31,486 raw indels and 72,516 raw single nucleotide polymorphisms (all but 17 of which are unsafe). The analysis error is the reason for the huge amount of mutations in Monroe.
The majority of Monroe’smutants are unassociated withhomomeric runs, but they are clustered within 10 base pairs of each other or unexpectedly common. The Weng HQ data has 2.5% clustered in it. Many of Monroe’s putative mutations are associated with more than one error: about 34% are associated with A/T homomeric runs and in a tight cluster. As centromeres are prone to mapping errors10, mis-mapping probably explains why 40.9% of LQ mutations are centromeric (see, for example, Extended Data Fig. 1e) compared with 27.9% in Weng HQ.
This neighbour base matching affecting both A and T in Monroe’s data is an expected bleed artefact with no biological basis. By contrast, we expect CpG>TpG mutations to be common given well-described methylated CpG hyperinstability15. In Monroe’s data, this is also the case.
Finally, to directly address the possibility that our conclusions reflect unknown sources of bias in inherently uncertain somatic calls, we reanalysed germline mutations from our study2 along with mutation accumulation experiment data generated in several independent studies (Supplementary Table 1). This meta-analysis of >10,000 germline mutations confirmed the previously reported, nearly universal reduction in single-nucleotide mutation rates in gene bodies, essential genes and regions marked by H3K4me1 (Fig. 2a–c; ref. The notable exception comes from plants lacking the mismatch repair protein MSH2 (Fig. 2a; ref. 5). A similar pattern is seen when somatic mutations were called with very stringent criteria in plants deficient for the MSH2 partner MSH6, using a tool specifically designed for rare somatic mutations14 (Fig. 2d). This was predicted by H3K4me1, which showed that MSH6 was attracted to gene bodies. The analyses of over 43,000 de novo germline alterations in rice also show that genes in the conserved genes, H3k4me1-marked regions and the salivary regions experience lower mutations rates.
The bleed-through errors that Wang and his colleagues think affect sequences up to five nucleotides away from homopolymers occur on Illumina platforms at positions immediately adjacent to a run of identical bases. Moreover, their simulation of sequencing errors apparently assumes that 100% of sequencing errors occur as a product of homopolymer bleed-through. Estimates of sequence errors only report 0.7 to 5.2% of the results as being Homopolymer bleed-through7. For high-quality germline calls, only 12.0% could be potential bleed-through errors, and on their own cannot explain the 50% reduction that we observed in gene.
The reported relationships between Epigenomic Features and Mutation Rates are well supported mechanistically. We agree that there are issues and inherent uncertainties with somatic mutation calling, which make it difficult to know the accuracy of individual calls in the very large set of loosely filtered somatic variants2. There is a proposal that the observed patterns are only the result of errors in the sequence of events.