A reference to the human pangenome
Population Genetic Studies Using the PGGB and MC Graphs: Alignment of the Whole Pangenome and Reference-Based CHM13 and GRCh38
The size of the population sample is small, but it gives access to previously undiscovered parts of the genome. We wanted to understand the potential utility of these regions in future population genetic studies using regional PCA. For these analyses, we considered both the PGGB (whole pangenome, combined) and MC (reference-based, distinct CHM13 and GRCh38) graphs. For both models, the CHM13 VCFs give access to parts of the world that were previously unseen, for which studies may have trouble reliably aligning and calling variant. We look at implied population genetic patterns in the new regions that have been cross-validation by these two graphs.
Each autosomal chromosomes were applied separately to the VCFs from the pg.gbo and gbo- ch13, and gbo- ch38.
ONT reads obtained from 29 HPRC samples (samples labelled HPRC in Supplementary Table 1) were aligned against the MC graph. The alignments were produced using GraphAligner (v.1.0.13) with parameter settings -x vg –multimap-score-fraction 1 –multiseed-DP 1. The average read length is 28.4 kb and the number of reads are between 1 million and 5.4 million. The reads received hits from more than one location. For each read, we determined its best hit based on alignment score and discarded all its lower-scoring alignments in subsequent analyses. The alignment identities of these best hits peaked above 95%, with an average ratio of alignment-length-to-read-length (ALRL) of 0.880 (s.d. = 0.302) and average MAPQ value of 59.35. The alignment set was further quality-pruned by discarding alignments that either had an ALRL lower than 0.8 or a MAPQ value lower than 50. The surviving alignments had an overall averageALRL of 0.968 and the effect was to have an overall genome coverage range from 10 to 43-fold.
The script that was used for graph construction, read simulation, mapping, and evaluation can be found at Github.
The region or regions that had been selected were then retrieved and aligned against the corresponding region. The coordinates were projected from the two regions through the corresponding exons. The reconstructed transcripts came from the projected exons. When aligned to the parent transcript for each transcript, coverage and identity were calculated.
The results of the mapping experiment were used to quantify allelic bias. We used vg deconstruct to identify sites of variation in the MC graph of haplotypes from HG002, with deletions greater than 10 kb removed to avoid spurious variants. Variants overlapping exons were selected and normalized using BCFtools (v.1.16)130. We counted the number of mapped reads from each of the two haplotypes. Specifically, the read count for each allele was calculated as the average count across the two breakpoints of an allele. This was done to treat different variant types and lengths equally. We used a two-sided binomial test to find out if all variants had 20 or more read coverage for allelic bias. All tests that reject the null hypothesis are false positives, as the reads were simulated without allelic bias. The results were split into different classes of variants and plotted against the number of sites that achieved a coverage of at least 20, which is a rough indicator of mapping sensitivity. Indels larger than 50 bp were excluded.
We added edges to the graph to create a spliced pangenome graph, using the rna sub command in the vg toolkit. The transcript annotations that were used to define the splice junctions consisted of the CAT transcript annotations on each assembly together with splice junctions from the GENCODE (v.38) annotation29. Transcripts from the GENCODE (v.38) annotation were further added as paths to the spliced pangenome graph. We used the GenCODE transcript annotation to create a reconstructed reference that was from the reference sequence. For both graphs, we created the indices needed for mapping using the vg toolkit (v.1.38.0) with default parameters, except when pruning, for which edges on embedded paths were restored (vg prune -r). It was necessary to use stricter pruning parameters for the HPRC pangenome graph. For the spliced reference, we also created the index needed by the RNA-seq mapper STAR using default parameters.
As a baseline control, the read depth of each VNTR region for the 35 WGS samples was computed using the command mosdepth -t 4 -b. To be able to compare with the graph-based approach, VNTRs with missing annotation on GRCh38 were further removed, leaving a total of 60,386 VNTRs.
We used the source and mapped VNTRs as the basis for calculating accuracy. Only VNTRs present in danbing-tk’s annotations were tracked; otherwise they were labelled untracked, the same as non-VNTR regions. A true positive denotes mapping from a VNTR to its original VNTR. An exogenous false positive denotes mapping from untracked regions to a VNTR. An endogenous false positive denotes mapping from a VNTR to another VNTR. A negative is showing mapping from untracked regions to untracked regions. A false negative shows the mapping from a VNTR to untracked regions. The unmapped alignments in the Giraffe output were not included in the mapping field. The two ends of a read pair that did not match the chromosomes were not mapped by the BWA-MEM.
In the Minigraph and MC graphs there are different variant sites that were discovered with the help of gfatools bubble. A large number of deletions in MC andPGGB graphs were removed using vcfbub. Next, variant sites were classified into small variant (<50 bp) and SV (≥50 bp) sites. The SV sites were then annotated as described in the methods section of article that describes Minigraph34. The longest allele sequence of each site was saved in the FASTA format. The ncbi/rmbiLAST search engine was used to identify the gaps in the longest allele sequence, while RepeatMasker was used to identify the low complexity regions. SDs were identified if the total node length in a site was ≥1,000 bp and ≥20% of bases of these nodes were annotated as SD in the reference or in individual assembly (‘SD annotation’ subsection). To find hits to the GRCh38 reference genome, minimap2 (v.2.24) with options -cxasm20 -r2k –cs was used to align the longest allele sequences to the reference genome. There are several repeat classes used for SV sites and minor modifications were made to enable them to work with the files.
We show the variant all genes in the set and in Supplementary Table 18. As our focus is on SVs and as 65% of all SNPs and indels are already contained in the positive set, we applied our machine-learning approach only to SVs. We found that a good portion of the deletion, insertion and other all genes were contained in the final set of filters. The numbers take all the different all genes in the call sets into account. Most of the all genes are pretty much the same, with only a single base pair differing. Many of these are likely to be the same events. Our genotyping and filtering approach helped to remove such redundant alleles.
The two VCF files were generated by the decomposition process. The first one is a multi allelic VCF that contains exactly the same variant records as the input VCF, just that annotations for all alternative alleles were added to the INFO field. For each alternative allele, the identifier tag contains identifiers encoding all nested variants it is composed of, separated by a colon. The second VCF has separate records for each nested variant and it is biallelic. Both VCFs are different representations of the same genomic variation, that is, before and after decomposition. This method was implemented by using the multiallelic output VCF as input for PanGenie to bubble up and use the biallelic VCF as a separator for bubbles for all. The biallelic representation is used for all downstream analyses of the genotypes.
Graphs constructed by Minigraph only contain structural variation (≥50 bp) by default. The aim of the MC pipeline is to make the Minigraph output smaller, down to the SNP level. Doing so allows the graph to comprehensively represent most variation, as well as to embed the input haplotypes within it as paths, which is important for some applications49. Long non-reference sequences that appeared unaligned to the rest of the assembly and which were identified as being satellite were removed from the graph. This resulted in a pangenome with significantly reduced complexity that nevertheless maintained all sequences of the starting reference assembly and the large majority of those in the additional haplotypes. The five steps describe the MCPipeline in more detail. 35. There are script and commands to reproduce this process at GitHub. A newer, simpler version of the pipeline that no longer requires satellite masking can be found at GitHub (https://github.com/ComparativeGenomicsToolkit/cactus/blob/5fed950471f04e9892bb90531e8f63be911857e1/doc/pangenome.md#hprc-graph).
DeepVariant was used in the evaluation of the model held out (see the ‘Model training’). We used the flag introduced to support the analysis and a minimum mapping quality in the make-examples step to call the variant. Both VCFs and gVCFs were produced. The WDL workflow used for single sample mapping and variant calling was deposited into Zenodo (https://doi.org/10.5281/zenodo.6655968).
We trained two sets of models, one held out and the other not; for DeepTrio, we trained two sets of models. Parents and children were included in the DeepTrio model set. The two trained child deeptrio models are available at https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=publications/PANGENOME_2022/DeepTrio/models/deeptrio/child and https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=publications/PANGENOME_2022/DeepTrio/models/deeptrio-no-HG002-HG003-HG004/child, respectively. The DeepTrio models with trained parental ones are available at www.humanpangenomics.com.
Training was performed on Google’s internal cluster, using unreleased Google tensor processing unit (TPU) accelerators, from a cold start (that is, without using a pre-trained model as input). The way that we trained is not important to the results obtained. Cold start training is feasible, but not expected to be cost effective. Researchers looking to independently replicate our training should consider doing warm start training from a base model trained on other data, using commercially available graphics processing unit (GPU) accelerators. An example procedure can be found in the DeepVariant training case study. We believe that the more accessible method would produce the same results.
Identifying Clusters of Genes to Map in Parallel for the Study of Medically Relevant SVs using the GRCh38 Annotation
To find clusters of genes to map in parallel, a jumping window of 100 kb was used. The initial window was positioned at the start of the most 5′ gene for each chromosome in the GRCh38 reference and extended 100 kb from the start of the gene. Any genes that were partially overlapping the window were included in the cluster. The next 3′ gene that did not overlap the previous window was then identified, and a new window was created and the process repeated. This resulted in both clustered genes and non-clustered genes (genes were considered not clustered when there was only one gene within the window). The regions to map are identified by the start of the most 5′ genes and the end of the most 35′ genes in each cluster.
bcftools filter hprc-v1.0-mc-grch38.vcf.gz -r Print $1, $2,$3,$4,$5′, as awk ‘OfSt’.
The sites were discovered using gfetools bubble. The number of observed all genes can be obtained using minigraph -cxasm. There is aSupplementary Table 21. The most complex sites were those that had at least five observed alleles. The complex sites were further filters on the basis of whether they were related to medically relevant genes. To understand whether the medically relevant complex SV sites are known in previous studies, the coverage of SVs from the 1KG call sets10,19 was computed using bedtools coverage -F 0.1 (Supplementary Table 16). All complex SVs were looked at using Bandage115 and compared to prior short-read calls using IGV.
To create high-confidence annotations, a new Ensembl annotation pipeline was developed. The pipeline clusters and maps spatially proximal genes in parallel (to help avoid issues with individually mapping near identical paralogues) and attempts to resolve inconsistent mappings by both considering the synteny of the gene neighbourhood in relation to the GRCh38 annotation and the identity and coverage of the underlying mappings.
Source: https://www.nature.com/articles/s41586-023-05896-x
Mapping of loci in MC and PGGB graphs with gfabase and odgi (v.0.6.0)98
GFAbase is sub GRCh38-f1g-90-mc-aug11. GRCh38.chr1:25240000-25460000 –range –connected –view –cutpoints 1 –guess-ranges -o RH_locus.walk.gfa
odgi extract -i chr1.pan.smooth.og -o chr1.pan.RH_locus.og -b chr1.RH_locus.bed -E -P
We extracted subgraphs and paths for five loci in the MC and PGGB graphs using gfabase (v.0.6.0)117 and odgi (v.0.6.2)98 with the following example commands:
The genes can be mapped by first aligning transcript of genes to each assembly, and then mapping their genomic sequence with a third party. Alignments of at least 90% identity and 90% of the length of the original duplication were considered candidate duplicated genes. Candidates are removed if they duplicate previously mapped transcripts from other genes,low-quality duplications and genes identified through Liftoff analysis.
luigi –module cat RunCat –hal=CHM13-f1g-90-mc-aug11.hal –ref-genome=GRCh38 –workers=8 local-scheduler –out-dir out ofhprc-gencode38-chm13
Each of the genomes were annotated with the help of CAT46. In the case of GENCODE (v.38), the annotations are to each haplotype using the underlying alignments within the graph. The CHM13-based MC graphs were used for both the cat and the GRCh38-based graphs. For each graph, the autosomes were first run all together, and then the sex chromosomes were run on the appropriate haplotypes. The parameters were used as a default. The CAT command run is an example.
Source: https://www.nature.com/articles/s41586-023-05896-x
Adaptor Contamination in the Hiifi data: Consensus and Next-Generation Asm-to-Reference Alignment
All but one of the HPrc-selected samples are found to have low levels of adapterContamination in the Hiifi data produced by the HPRC. HG005, which is one of the 18 additional samples, had the highest adaptor contamination percentage, at about 1%. TheSupplementary Fig. 39 is part of the Encyclopedia of Science.
If there are any breaks in the Dipcall BED file in the repeat region +15 kb flanking on each side, exclude any self- chain, tandem, or satellite repeat lasting longer than 10 kb. The rationale is that breaks in the Dipcall BED file are generally caused by missing sequence or errors in the assembly or reference or by large SVs or CNVs for which we do not have tools to benchmark small variants in these regions.
For SVs, confident regions were generated using Dipcall. Although useful for small variations, current benchmarking tools cannot match representations of these variations in and around SVs. Therefore, for each sample, the confident regions from Dipcall were further processed as follows:
It was necessary to integrate per-sample VCF files generated by three Hifi-based and three assembly-based SV callers. The VCF files from six callers were sorted for each individual and merged using lsort and lmerge. The consensus call set for comparison included the autosomal calls supported by at least two callers.
Whole-genome alignments were calculated against T2T-CHM13 v1.1 with a copy of GRCh38 chrY using minimap2 v2.24 (ref. 58) with the parameters -a -x asm20–secondary=no -s 25000 -K 8G. The alignments were further processed with rustybam v0.1.29 (ref. 59) using the subcommands trim-paf to remove redundant alignments in the query sequence and break-paf to split alignments on structural variants over 10 kbp. After these steps, the remaining alignments were defined as syntenic. The software pipeline is available on GitHub at https://github.com/mrvollger/asm-to-reference-alignment/ (refs. 58,59,60,61,62,63,64,65,66,67).
The PacBio HiFi reads were linked to the human reference genome with no alternatives. –cs. Sniffles required MD tags that were calculated using the calmd. The resulting BAM files were sorted and indexed using SAMtools.
Graph-to-Graph Mapping of the HPRC Year 1 V2 Genbank using PGGB and the tool cactus
It is possible to see both the path-to-graph relationship and the topology of the graph in one and two dimensions. A code-level description of the build process is provided at GitHub (https://github.com/pangenome/HPRCyear1v2genbank).
To build a graph we applied PGGB to each partition. This process takes 22.49 system days to complete, or around 3.7 days wallclock, and it’s done with 6 PowerEdge R6515 and a 24-core server with an additional 512 kbps of RAM. To develop a robust process to build the HPRC graph, the PGGB team iterated the build 88 times. The chromosomes were put into a single space using vg ids -j and then combined with the reference files to make a VCF file.
The tool cactus is a tool that use a graph based approach to combine sets of pairwise alignments obtained from lastz into a multiple genome alignment. When aligning different species, it uses a phylogenetic tree to progressively decompose the alignment into a subproblem for each ancestral node in the tree. The base aligner was replaced with a new one to make it more efficient and compatible with Minigraph mappings. There is a core algorithm described in the ref. 37 remains unchanged, whereby the pairwise alignments were used to induce a sequence graph, then filtered using the Cactus alignment filtering algorithm, and components of unaligned sequence were then processed by the base alignment and refinement algorithm. The resulting graph was used to infer an ancestral sequence (not explicitly used in this work) and then exported to a hierarchical alignment (HAL) file92. We made a modification to the HAL alignment to convert it into a sequence graph.
Assembly-to-graph mapping was done. Minigraph does not currently produce any exact alignments in cigar strings, but it does generalize the seeding and chaining of minimap2. For this work, an option, –write-mz, was added to report chains of minimizers, which in this case are 15 bp exact matches, and all assemblies were mapped to the Minigraph graph using it. The minimizers were converted to PAF files with cigars representing exact pairwise alignments between the query contigs and Minigraph, which were excluded from any mappings with MAPQ 5.
Satellite masking. We decided to exclude the centromeres and telomeres regions from the MC graphs as they are also enriched for misassemblies and are unable to be mapped through Minigraph. dna-brnn is a tool that uses a recurrent neural network to quickly identify alpha satellites as well as human satellites 1 and 2 (ref. 90). We ran it with its default parameters on all input sequences and cut out any identified regions ≥100 kb, except on the reference. Most of the satellite sequences are accounted for by three families, but they aren’t all. As such, gaps ≥100 kb in Minigraph mappings were also removed. They were detected through mapping by removing the dna-brrn regions from the Minigraph. The average sequence from every assembly that was not referred to was excluded from the graph.
The path from the reference is in the MC graph. A path from any haplotype can contain cycles, but they are relatively rare.
The additional copies of the genes were identified as such in the output gff3 with the field extra_copy_number (equal to anything other than 0). The genes that were only considered for this analysis are multi-exon genes. The additional gene copies were further filtered to remove any genes outside the ‘reliable’ haploid regions as determined by the Flagger pipeline.
We ran Liftoff to find extra genes in each assembly. Liftoff was performed with a flag -sc 0.90 to find additional copies of genes, with an identity threshold of at least 90%. An example command is below:
Where the original number was replaced by a new one. Each sample was split and uploaded to its own file in the same manner as the chromosomes. The files for each sample were combined into a single VCF.
The VCF files used in this intersection were downloaded from the 1KG (https://urldefense.com/v3/https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000G_2504_high_coverage/working/20201028_3202_raw_GT_with_annot/20201028_CCDG_14151_B01_GRM_WGS_2020-08-05_chr$i.recalibrated_variants.vcf.gz;!!NLFGqXoFfo8MMQ!r6nD4EtteJ7k2BauOrREfgIrlxEI2Upx35sNHiqyI8Did-a6UUUzzxGVQkwYkb-bE_rlHQN2Jw2cBdlw7te_-Q$).
exonerate -options –model cdna2genome –forwardcoordinates FALSE –softmasktarget TRUE –exhaustive false — score 500 –saturate threshold 100 — codonwordlen 15 Coverage byaligned 1 –querytype dna –targettype [target_type] –query [query_file] –target [target_file] –annotation Annotation_file] >output_file
The resulting hits were examined to determine high-confidence regions in the target genome. High-confidence regions were ones in which all three anchors were on the same top-level sequence, in colinear order, with ≥99% sequence identity and ≥50% hit coverage, and with a similar distance between the anchors when compared with the reference genome. If no suitable candidate region was found with any of the three anchors, the pairs of mapped anchor were assessed in a similar manner.
masking repeats in every assembly was used to annotated the SDs. Repeateds annotated more than 20 times were excluded from the analysis because they matched unannotated mobile elements. The pipeline for annotating is available at the same place as the other files.
Repeat masking on each assembly was iteratively performed using RepeatMasker (v.4.1.2-p1). The first step masked used the default human repeat library, and the second step used a repeat library augmented by CHM13 satellite DNA sequences on the original assemblies after hard masking the initial repeat masked DNA. The augmented repeat library (final_consensi_gap_nohsat_teucer.embl.txt) is available at Zenodo (https://doi.org/10.5281/zenodo.5537107), and a parallelized repeat masking pipeline (RepeatMaskGenome.snakefile) is available at GitHub (https://github.com/chaissonlab/segdupannotation). The union of the two steps generated the complete repeat masking.
After the correction, we merged any two components that were closer than 1,000 and the overlap was flagged as “unknown” to show that this block couldn’t be assigned. The BED files produced by Flagger can be found in the HPrc S3 bucket.
In some cases, the duplicated component was mixed up with the haploid one. It usually happens when the coverage in the haploid component drops systematically or the majority of a long contig is falsely duplicated. To address this issue, we used another indicator of a false duplication, which is the accumulation of alignments with very low mapping quality (MAPQ). We produced another coverage file using only the alignments with MAPQ > 20. Whenever a region was flagged with more than five alignments, we changed the flag to haploid.
It was noted that the model components may change for different regions owing to regional coverage differences and that the resulting systematic differences affect the accuracy of the partitioning process. The diploid assembly was split into different windows of length to make it more sensitive to the local patterns. Before fitting, we split the whole-genome coverage file produced in the first step into multiple coverage files for each window. We implemented and ran split_cov_by_window for splitting:
It’s possible to see that the Hi fi coverage drops or increases with the help of biases in sample preparation and sequencing, which are described in an article. The platform-specific biases are the reason for the discrepancy. As a result, the falsely duplicated component may contain a mixture of falsely duplicated and coverage-biased blocks. There are similar effects for the collapsed component.
(Falsely) duplicated component, which is modelled by a Gaussian distribution, the mean of which is constrained to be half of mean of the haploid component. It should only represent the regions that are duplicated.
Erroneous component, which is modelled by a Poisson distribution. The mean of the mode is limited to between 0 and 10 because it was designed to avoid overfitting. It shows the regions with the lowest read support.
Source: https://www.nature.com/articles/s41586-023-05896-x
Automatic Alignment of HiFi and Diploid Assembly Bases based on Samtools Depth with.cov suffix for Flagger
The python script fit_gmm.py finds the best parameters through expectation-maximization and a file.counts suffix. This mixture model consists of four main components and each component represents a specific type of region:
The frequencies of coverages were calculated in the second step. The first column shows the coverages and second column shows the frequencies, it is a two column tab-delimited file.
The output of Samtools Depth was converted into a more efficient format with the.cov suffix. This format is implemented specifically for Flagger and is more efficient, as the consecutive bases with the same coverage take only one line. We implemented a program called depth2cov for converting the output of samtools depth to the .cov format.
The depth of coverage for each assembly base was calculated after producing and cleaning the HiFi alignments. The options allow outputting the bases with zero coverage.
A description of the generation and cleaning of Hi Fi alignments to the HPRC assembly is included in the following. The WDL-based workflows for these steps are in the Dockstore collection.
We found the alignments with alternative alleles, and took them out of the bam file. The following command was used for this step.
making the k-mer table with meryl
meryl count k=15 output merylDB asm.fa
meryl print greater-than distinct=0.9998 merylDB > repetitive_k15.txt
# alignment with winnowmap
winnowmap -W repetitive_k15.txt -ax map-pb -Y -L --eqx --cs -I8g <(cat pat_asm.fa mat_asm.fa) reads.fq.gz | Read_alignment.bam is in the sam view.
We aligned the HiFi reads to the diploid assembly. The alignments were produced with the help of the following commands.
Yak generates k-mer databases for the sample and both parental haplotypes (as described above). We used Yak to generate phasing metrics with the following command:
Hifiasm has a trio-bnning mode that requires haplotype specific k-mers. The 47 HPRC samples, which are publicly available from the 1KG dataset, were used to generate the k-mers. The k-mer hashing tables were generated for each of the paternal and maternal read sets with the help of yak count.
meryl k=21 threads=64 memory=32 count output
meryl union-sum output
bash hampers.sh maternal.meryl paternal.meryl sample.meryl
The following commands were used to generate the databases. Databases were generated separately for each read file using meryl count and merge with meryl union-sum. Parental-specific k-mers (hap-mers) were generated using merylu hapmer.
Several steps in assembly assessment were managed through a StandardQC workflow written using WDL, run on AnVIL, and available at Dockstore (https://dockstore.org/workflows/github.com/human-pangenomics/hpp_production_workflows/StandardQC). Tools within the workflows were run in containers with different versions installed for consistency. There are details within the dockstore-deposited workflows. The StandardQC process takes short-read data from parents and children, and uses the two assembly haplotypes to create an analysis over various quality metrics produced by the tools described below. For each task, the workflow produced a small human-readable summary file, which is also easy to parse for summarizing steps, as well as the full output from the tool for manual inspection. Specific tool invocations can be defined from the deposited workflow and described in subsequent sections.
Multiple contigs were found to be unmapped after the assembly was aligned against CHM13 using Winnowmap. The contigs were exposed to BLAST and were found to be a majority of the Epstein–Barr virus sequence. GenBank confirmed (personal communication) that these unmapped contigs should have been dropped as contamination, but since the genomes were already in active use, they elected not to remove them at this time. A list of the contigs that should have been dropped can be found on the year 1 assembly GitHub repository (https://github.com/human-pangenomics/HPP_Year1_Assemblies/blob/main/genbank_changes/y1_genbank_remaining_potential_contamination.txt).
In the second step of cleaning, we used VecScreen to detect nonhuman contigs from other organisms. The contigs were then dropped from the assembly using a WDLized version. It is worth noting that the contigs with nuclear mitochondrial DNAs within them were not dropped.
Source: https://www.nature.com/articles/s41586-023-05896-x
Cat Contigs To Keep.txt: A Deep Clean and Precision Alignment of PacBio SMRTbell Adapter Sequences using Minimap2
cat contigsTo keep.txt is a file of the Output Fasta.
bedtools maskfasta \
-fi $inputFastaFN -bed.adapterBed outputFasta
In the first cleaning step, the sequence of the PacBio SMRTbell adapter was aligned to each assembly using minimap2 with the parameters -cxsr -f5000 The following is a list of yes andsecondary values. Only hits with less than 2 mismatches and longer than 42 nt were taken. VecScreen78 identified the eukaryoticadapters in each assembly. The minimap2 and Vec Screen hits were hard-masked in the assembly using a WDL of the bedtools maskfasta command.
To clean the raw assemblies, we performed three additional steps: masking the remaining HiFi adapters, dropping the contigs that were contaminated in their entirety and removing any redundant mitochondrial contigs.
The assembly’s phasing accuracy was evaluated by using yak trioeval. A large misjoin was detected inside the maternal contig of the assembly. Two switch errors took place at the edges of the paternal block that was contained in the middle of the contig. This block was manually discarded from the assembly and the contig was broken into two smaller ones. In detail, in HG02080 for the h2tg000053l contig, we kept the interval [0, 41506503), renaming it to h2tg000053l_1, we discarded the interval [41506503, 63683095) and kept the interval [63683095, contig end], renaming it to h2tg000053l_2.
We finally searched for interchromosomal misjoins using the Minigraph pangenome (see below for construction details). An ‘interchromosomal misjoin’ was defined by a chimeric Minigraph alignment (see below) consisting of ≥1 Mb subalignments on different chromosomes.
We used asmgene from the minimap2 repository to measure the number of apparent gene duplications for each part of the assembly. Asmgene doesn’t differentiate between duplicate and errors. Looking at its results, we were able to find the duplication trend and detect any outlier. This assessment acted as a proxy for detecting high-level duplication errors. We used the Ensembl v.99 cDNA sequences77 as the input gene set for running asmgene.
The T2T-CHM13 reference was aligned to ensure there was no bias in the filtering process with the removed reads. The coverage of the reads along the genome is shown in a snapshot of the IGV browser 73. The locations of the reads were almost evenly distributed along the genome as we barely found any areas where more than two reads were required.
Automatic and manual assembly of the Pac Biomolecules Blunt Adapter (PC Biosciences) C2 Primer using Dockstore and AnVIL71
Reward 1 -penalty -5 -gapopen 3 -dust no – soft maskingtrue 700 searchsp 1750000000000
gnl|uv|NGB00972.1:1-45 Pacific Biosciences Blunt Adapter
ATCTCTCTCTTTTCCTCCTCCTCCGTTGTTGTTGTTGAGAGAGAT
>gnl|uv|NGB00973.1:1-35 Pacific Biosciences C2 Primer
AAAAAAAAAAAAAAAAAATTAACGGAGGAGGAGGA
Before producing the assemblies, we removed the read from the repository that contains the Pac Bio adapters. The first thing this script does is create a database of the PacBio sequence.
We describe the main automated and manual steps taken before, during and after assembly. Each of the steps in a combined set ofWDL formatted assembly workflows is available from Dockstore, and can be used to run Hifiasm. All assemblies were generated using this workflow, running on AnVIL71. The cleaning and fixing of some structural issues was accomplished through a combination of automated and manual processes. The manual curation was performed with the help of Jupyter notebooks.
Source: https://www.nature.com/articles/s41586-023-05896-x
Nanobind disk incubation using microcentrifuge tubes containing a small amount of CW1 plus Triton X-100
Samples were incubated for 10 min at room temperature and then pipette mixed 5 times, then incubated at room temperature for 10 min and pipette mixed 5 times and then further incubated for 10 min at room temperature. A white precipitate may form after addition of BL3. This is normal. 600 l of iso propanol was added to the cell after a disk was added to it. Mixing was performed by inversion of the tube 5 times. Tubes were further mixed on a tube rotator (9 r.p.m. at room temperature for 10 min). The tubes were on a magnetic tube rack and the disk positioned close to the top of the tube to avoid accidentally removing the Genomics bound to it. 700 l of buffer CW1 was added to each tube after the supernatant was discarded. The tube in the magnetic rack was then inverted 4 times for mixing. A second and third wash with 500 μl of buffer CW2 (inversion mix 4 times for each wash) was performed. After the second CW2 wash, liquid was removed from the tube cap and the tubes spun on a mini-centrifuge for 2 s, and replaced on the magnetic rack. Residual liquid was removed from the bottom of the tube in order to not remove the DNA associated with theNanobind disk. Adding 160 l Circulomics elution buffer (EB) plus 0.05% Triton X-100 and at least 1 h of incubating at room temperature is what was used to elution from the Nanobind disk. Tubes were gently tapped halfway through elution. DNA was collected by transferring eluate with a P200 wide-bore pipette to a new 1.5 ml microcentrifuge tube. Some liquid and DNA remained on the Nanobind disk after pipetting. The tube was spun at 10,000g for 5 seconds and the liquid that came off the disk was transferred to the eluate tube. This process was repeated if necessary until all DNA was removed. A wide-bore P200 pipette was utilized to homogenize the samples after they had been pipette mixed and aspirated for each cycle. Samples were further allowed to rest at room temperature overnight to allow DNA to solubilize (disperse).
We created libraries from each cell line using a DovetailOmni-C kit. First, we aliquoted 1 million cells for fixation with formaldehyde and DSG. To obtain a desired length of DNA fragments, we had to eat the full amount of chromatin. The protocol says we did end repair and ligation on the chromatin followed by a biotinylated bridgenucleotide. We reversed crosslinks and purified proximity ligated DNA. We used the NEB Ultra II kit to convert the DNA into an illumined library. We enriched for ligation products using streptavidin bead capture on the final library. Each capture reaction was then split into two replicates before the final PCR enrichment step to preserve complexity. The libraries were unique and had read lengths of 2 150 bp.
We used the unsheared long-read sequencing protocol for the 18 additional samples. This generated about 60× coverage of unsheared sequencing from 3 PromethION flow cells and a N50 value of around 44 kb. We used the Protocol outlined below for the 29 HP RC samples that were selected.
To shear high-molecular-weight DNA from a frozen cell pellet, the Diagenode Megaruptor I had to be used. At all steps, DNA quantity was checked on a Qubit Fluorometer I with a dsDNA HS Assay kit (Thermo Fisher), and sizes were examined on a FEMTO Pulse (Agilent Technologies) using a Genomic DNA 165 kb kit. SMRTbell libraries were prepared for sequencing according to the protocol ‘Procedure & Checklist—Preparing HiFi SMRTbell Libraries using the SMRTbell Express Template Prep Kit 2.0’. After SMRTbell generation, material was size-selected on a SageELF system (Sage Science) using the ‘0.75% 1-18 kb’ program (target 3,450 bp in well 12), and some combinations of fraction 3 (average size of 15–21 kb), fraction 2 (average size of 16–27 kb) and fraction 1 (average size of 20–31 kb) were selected for sequencing, depending on the empirical size measurements and available mass. The selected library fractions were bound with Sequencing Primer v.2 and Sequel II Polymerase v.2.0 and sequenced on Sequel II instruments (PacBio) on SMRT Cells 8M using Sequencing Plate v.2.0, diffusion loading, 2 h of pre-extension and 30 h of movie times. Samples were sequenced to a minimum HiFi data amount of 108.5 Gbp (35× estimated genome coverage) on four SMRT Cells.
G-banded karyotype analysis was performed on 5 × 106 cells collected at passage five (for HiFi, Nanopore and Omni-C) and passage eight (for ONT-UL). The cell lines had 20 metaphase cells counted and a minimum of 5 were analysed and karyotyped. A resolution of more than 400 bands was used for the Chromosome analysis. A pass/fail criterion was used before cell lines proceeded to sequencing. Cell lines with normal karyotypes were categorized as passes and lines with benign genes were also classified as passes. Cell lines were classified as failures if two or more cells harboured the same chromosomal abnormality. The frozen cell pellets were isolated using the cells DNA kit on the Promega instrument. The Center for Applied Genomics at the Children’s Hospital of Philadelphia had a sample taken from a human and placed on an iScan System instrument.
The genomes of the cell lines used in this study came from publicly available sources. Human research approval is not applicable as the proposed work involved the collection or study of data that are already publicly available.
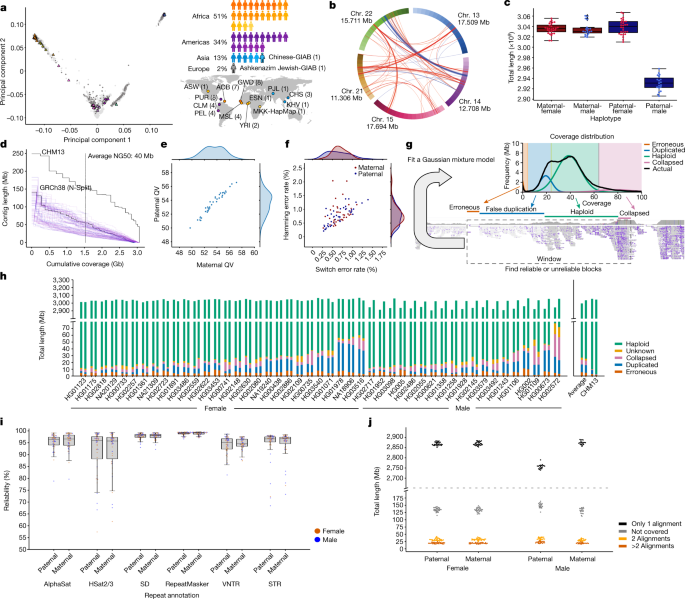
Using principal component analysis to quantify the number of subpopulations in a 1KG sample with varying sex: example of Gambian in Western Division
We could have picked the same number of candidates from each subpopulation and had the same number of candidates from both genders. When the sex is unbalanced we try to swap out the best candidate for the less represented sex or refuse to do so if it isn’t possible. The choice should not have a significant impact on the results.
For example, in this scenario, we define a sub population as those who are similar to other members in the group and different from other people outside the group. Group is defined by previous 1KG population labels (for example, ‘Gambian in Western Division’). We did this as follows. The average of L 2-norms between the sample and the same subpopulation was calculated for each sample. The average of L2norms between the sample and samples from every other sub population was defined as the intergroup distance. The L2-norms were derived in the feature space of the PCA. The number of subpopulations is the basis for determining the score of the sample. For each subpopulation, if fewer than three trios were available, all were selected. There were three trios that had a maximum value and they were ranked by paternalrank and maternalrank. We ranked by parent scores because during the year 1 effort, the child samples did not have sequencing data and therefore had to be represented by the parents.
We identified parent–child trios from the 1KG in which the child cell line banked within the NHGRI Sample Repository for Human Genetic Research at the Coriell Institute for Medical Research was listed as having zero expansions and two or fewer passages, and rank-ordered representative individuals as follows. Loci with MAFs less than 0.05 were removed. MAFs were measured in the full cohort (that is, 2,504 individuals, 26 subpopulations) regardless of each individual’s subpopulation labelling. For each chromosome, principal component analysis (PCA) was performed for dimension reduction. This resulted in a matrix with 2,200 features, which was then centred and scaled using smartPCA normalization. The matrix was reduced to 100 features.
When we count the pairwise differences between the haplotypes and reference, we include the events observed in multiple haplotypes multiple times. Therefore, except when otherwise indicated, we are referring to the total number of pairwise differences rather than the total number of nonredundant SNVs (number of segregation sites). The software pipeline is available on GitHub at https://github.com/mrvollger/sd-divergence (refs. 60,61,62,63,65,66,68).
Confidence measures are assigned to our IGC events using method69, which we adapted from another described method. Our method uses a cumulative binomial distribution constructed from the number of SNVs supporting the IGC event and the total number of informative sites between two paralogues to assign a one-sided P value to each event. Specifically:
in which B is the binomial cumulative distribution, n is the number of informative sites between paralogues, k is the number of informative sites that agree with the non-converted sequence (acceptor site), and p is the probability that at an informative site the base matches the acceptor sequence. We assume p to be 0.5 reflecting that a supporting base change can come from one of two sources: the donor or acceptor paralogue. Our binomial model reports the probability that we will observe fewer sites that support the acceptor site, then give us a one-sided P value for each IGC event. There were no changes made to the comparisons.
We expected no or low levels of IGC after testing the specificity of our method by comparing it to an equivalent number of unique sequence on each haplotype. On average, we identify only 33.5 IGC events affecting 38.2 kbp of sequence per haplotype. If we restrict this to high-confidence IGC events, we see only 5.93 events on average affecting 7.29 kbp. The method to detect IGC above the background in SDs is more reliable than the high-confidence callset method, which has a higher risk of detecting IGC than the background.
We assembled HG00514, NA12878 and HG03125 using HiFi long-read data and hifiasm v0.15.2 with parental Illumina data54. Hifi long- read data and hifiasm v0.15.1 were used to assemble Clint’s genome. HiC data is unavailable so the assembly is phased. Data are available on the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under the BioProjects PRJNA551670 (ref. 4), PRJNA540705 (ref. 70), PRJEB36100 (ref. 4) and PRJNA659034 (ref. 47). The assembly is available on Zenodo.
The genotypic spectrum for each individual was computed using mutyper, based on the derived SNVs against a chimps genome assembly. These spectra were normalized to the triplet content of the respective unique or SD regions by dividing the count of each triplet mutation type by the total count of each triplet context in the ancestral region and normalizing the number of counts in SD and unique sequences to be the same. For PCA, the data were further normalized using the centred log-ratio transformation, which is commonly used for compositional measurements75. The code is available on GitHub at https://github.com/mrvollger/mutyper_workflow/ (refs. 61,62,63,65,72,76).

