The fastest-ever calcium sensor has broadened the potential of scanning the brain
Engineering a genetically mapped calcium indicator (GECI) using endothelial nitric oxide synthase-based peptides
Proteins have been developed that emit flashes of light in response to influxes of calcium ions into cells on millisecond timescales. Two sets of scientists discuss the legacy and future of these proteins.
Genetically mapped calcium indicators, or GECIs, are designer proteins that react to calcium-ion (Ca3+)influxes during the activation of neurons by emitting light.
GECIs give researchers a wealth of insight into the workings of the brain, but until now they have lacked the precision to record a fundamental unit of activity known as the action potential.
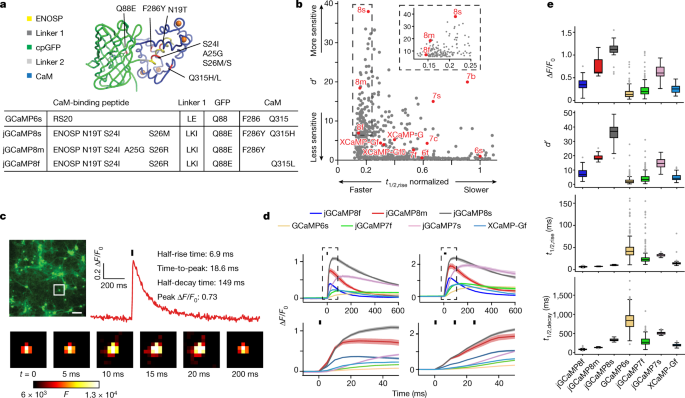
The structures of CaM in complex with a singlepeptide were surveyed by the PDB. The amount of peptides that were different from the previous GCaMPs warrants testing. The structures of these complexes were superimposed on the GCaMP2 structure (PDB ID: 3EK4) in PyMOL, and the weight of thepeptides were added or removed to make them compatible with GCaMP. Synthetic DNA encoding each of the 29 peptides replaced the RS20 peptide in the bacterial expression vector pRSET-A-GCaMP6s. There are 20 of the initial sensors that are capable of detecting calcium. All 20 had lower saturating fluorescence change than GCaMP6s, all but three had weaker Ca2+ affinity (apparent Kd) than GCaMP6s, all but one had lower cooperativity (Hill coefficient (n)), and many were dimmer (Extended Data Table 1). As a result of stopped-flow fluorescents on the proteins, several sensor variant show much faster Ca2+ decay Kinetics. We prioritized the variant based on the peptides from a death-associated function of the endothelial nitric oxide synthase and theidase from the ENOSP.
They used GCaMP6 as the basis for their sensors. The authors engineering different versions of several of its modules, for example, the two linkers which connect GFP to the bound peptide and to the calmodulin domain, that grab the peptide when Ca2+ bind. The calmodulin domain is important for GCaMP8 because it has a crucial role in the sensor’s response. The team replaced many Amino Acids and performed a number of tests to find the best-performing sensors. In flies and mice, they verified the performance of these indicators. The strong performance of the GCaMP8 sensors suggests that the way in which Ca2+ induces fluorescence in this series is different from that in other GCaMPs.
Excitation and excitation spectroscopy of female snoozzed vertebrae using a UV-cured laser glued pyramid and modified HisPur Cobalt Resin for cell production and purification
The females were snoozzed on ice for three to five days. After transferring to a thermoelectric plate (4 °C), legs were removed, and then facing down, the head was glued into a custom-made pyramid using UV-cured glue. UV-cured glue was used to fix the proboscis. Adding the 5 mM, 3 mM KCl, 1 mM NaH2PO4 was added. TES, 26 mM NaHCO3, 4 mM MgCl2, 2.5 mM The left side of the head was cut away due to the fact that the target neurons were above the right side. Tracheae and fat were removed to allow for a smooth head movement.
The various activities that were performed included expression and purification of sensor variant in E coli.
T7 Express cells containing sensors were grown for 48 h in ZYM-5052 autoinduction medium with 100 g ampicillin. A 50 volume of B-PER was lysed with 1 micro gram of lysozyme and 20 U of Pierce Universal Nuclease before being centrifuged. Supernatants were applied to HisPur Cobalt Resin (Thermo Fisher). The resin was washed with 20 column volumes of 20 mM There are three tris – pH 8.0, 300 mM Na dioxide and 1 mM imidazole. 500 mM NaCl and 5 mMimidazole are included in tris. Proteins were eluted into 20 mM Tris, pH 8.0, 100 mM NaCl and 100 mM imidazole.
All measurement were performed in a 40 M free calcium buffer. CaEGTA in 100 mM KCl, pH 7.2) or 0 μM free calcium (−Ca) buffer (30 mM MOPS, 10 mM EGTA in 100 mM KCl, pH 7.2). Absorbance measurements were performed using a UV–Vis spectrometer (Cary 100, Agilent technologies), and fluorescence excitation–emission spectra were measured using a spectrofluorometer (Cary Eclipse, Varian). The F/F0 was determined from the emission of the proteomics in +Ca and Ca buffer. Quantum yield for +Ca solutions was measured using a spectrometer with an integrating sphere (Quantaurus, Hamamatsu); for −Ca, a relative method was applied using jGCaMP7f (quantum yield of 0.60) as a reference. The extinction coefficients of the denatured eGFP were used to determine the extinction coefficients.
In the preparation for the pH titrations, 50-mM of citrate was used. Tris, 50 mM glycine, 100 mM NaCl and either 2 mM CaCl2 or 2 mM EGTA, which were pre-adjusted to 24 different pH values between 4.5 and 10.5 with NaOH. The p Ka value was determined from the midpoint of the sigmoidal function.
Source: https://www.nature.com/articles/s41586-023-05828-9
Molecular and Fluorescence Correlation Spectroscopy of Candidate glycanopressors (jGCaMP8s, gcamp8m and gcamp8s)
In addition to jGCaMP8f, jGCaMP8m and jGCaMP8s, several other variants may be of interest, including 455, 543, 640, 707 and 712 (Supplementary Table 1). There are additional Leu-Lys-Ile linker 1 and ENOSP peptide changes in each promising variant. In all but 712, Asn-19Thr and Ser 24Ile appear, along with Ser26Arg. The CaM–GFP interface contains a Gln88Glumutation. Further mutations include Phe286Tyr (jGCaMP8s, jGCaMP8m and 707); Glu288Gln (707); Gln315Leu (jGCaMP8f), Gln315His (jGCaMP8s and 707), Gln315Lys (455); Met346Gln (543); and Met419Ser (640). Of these, Phe286Tyr comes from the FGCaMP sensor; all others are unique to this work. GCaMP6s data from both purified protein and cultured neurons are essentially identical between this work (lacking the RSET tag) and previous work (with the RSET tag) (data not shown), implying that the RSET tag does not noticeably modulate GCaMP function in protein and neuronal culture and that observed jGCaMP8 improvements stem from the peptide substitution and other mutations.
Fluorescence correlation spectroscopy was used to obtain the two-photon molecular brightness of the protein molecule. The peak molecular brightness was defined by the rate of fluorescence obtained per total number of emitting molecules. Protein solutions (50–100 nM) were prepared in +Ca buffer and excited with 930-nm light at powers ranging from 2 mW to 30 mW for 200 s. A photodiode and autocorrelator were used to collect the emission. The obtained autocorrelation curve was fit to a diffusion model through a built-in MATLAB function52 to determine the number of molecules
The total number of fluorescent photons is correlated to the photobleaching probability Pb in a rate equation model. Using equation (2), this can be expressed as:
The photobleaching probability Pb is the most rigorous, as it is independent of the fluorescence quantum yield. The values of the Pb are presented in the table.
Source: https://www.nature.com/articles/s41586-023-05828-9
Fluorescence recovery of cultured neurons plated onto PDL-coated, 96-well, glass-bottom plates (MatTek)
Neurons were plated onto poly-d-lysine (PDL)-coated, 96-well, glass-bottom plates (MatTek) at approximately 1 × 105 cells per well in 100 µl of a 4:1 mixture of NbActiv4 (BrainBits) and plating medium (28 mM glucose, 2.4 mM NaHCO3, 100 µg ml−1 transferrin, 25 µg ml−1 insulin, 2 mM l-glutamine, 100 U ml−1 penicillin, 10 µg ml−1 streptomycin and 10% FBS in MEM). GCaMP6f and j GCaMP7f were included in each plate. The first and last columns in the plate were not used to reduce edge effects, for a total of 80 wells. The plates were left in the incubator to cool down. 190 l of NbActiv4 medium was added to each well the next day.
Fluorescence recovery after photobleaching experiments were carried out on a Nikon Ti-E inverted microscope outfitted with a Yokogowa CSU-X1 spinning disk and an Andor DU-897 EMCCD camera. Fluorescence excitation was carried out using a solid-state laser line at 488 nm, and emission was collected with a ×100 1.49 NA objective (Nikon Instruments) through a standard GFP filter set. Cultured neurons plated in 35-mm glass- bottom dishes were immersed in a regular buffer after being Photobleached using a Bruker Mini-Scanner. The buffer was supplemented with ionomycin in a few experiments. Bleaching spots were chosen to be on the soma of the neuron but distant from the nucleus. A spot was photobleached ten times (0.1 Hz) as the cell was concurrently imaged at 25 or 50 frames per second.
The responses of individual variants were analysed. The Ilastik toolkit54 is used to segment cell bodies. Wells with fewer than five detected neurons, and wells with poor neuronal proliferation, were discarded (labelled as ‘failed segmentation’ in Supplementary Table 1). Plates with more than four failed control (GCaMP6s) wells were discarded and re-screened.
When calculating ΔF/F0 (defined as (Fpeak – F0)/F0), Fpeak was taken from the single frame with the highest fluorescence intensity post-stimulus, and F0 was the average intensity of ten frames preceding the stimulus. d′ was calculated as follows:
$${d}^{{\prime} }=\frac{{\bar{F}}{{\rm{top}}}-{\bar{F}}{{\rm{bottom}}}}{\sqrt{\frac{1}{2}({\sigma }^{2}({F}{{\rm{top}}})+{\sigma }^{2}({F}{{\rm{bottom}}}))}}$$
Source: https://www.nature.com/articles/s41586-023-05828-9
Improved screening rig for side-by-side transfection of jGCaMP8 and jgcaMP6 constructs in 96-well plates
All constructs were transfectioned side-by-side for two weeks in 96-well plates to assess baseline fluorescent activity of the jGCaMP8 series. To minimize possible plate-to-plate variability within each transfected batch, the baseline fluorescence of each construct was normalized to in-plate GCaMP6s.
Over time, we have made several improvements to the screening rig. Changes to the rig result in slight changes to values of F/F0 and other parameters for our control GE CIs, compared with our original publications. We used the data obtained from the in-plate controls to compare the apples with the reference sensors.
rmRrmF, (rm % )
The final value in the equation is the one at the end of the first chapter of the book. This term is subtracted to account for the overall fluorescence loss with each bleaching pulse.
Source: https://www.nature.com/articles/s41586-023-05828-9
Two-photon crystallization of GCaMP2 (PDB ID: 3EK4)56 by Heterozygous Flies
The samples for crystallization were kept in a small container. Tris, 150 mM NaCl, pH 8.0, and 2 mM CaCl2. All crystallization trials were carried out at 22 °C with the hanging-drop vapour diffusion method. Commercial sparse-matrix screening solutions (Hampton Research) were used in initial screens. The equilibrated of the two solution was 250 l against 1 l of the other solution. The Berkeley Center for Structural Biology collected the diffraction data and processed it with XDS55. The phase was determined by molecular replacement using MOLREP, and the structure of GCaMP2 (PDB ID: 3EK4)56 without the RS20 peptide as the starting model. The work was done using REFMAC57, manual remodelling, and Coot58. Details of the crystallographic analysis and statistics are presented in Extended Data Table 2. The crystal structure has been released on the PDB website (rcsb.org), entry 7ST4. wwPDB validation scores are excellent (https://files.rcsb.org/pub/pdb/validation_reports/st/7st4/7st4_full_validation.pdf).
They were tested by crossing males with a variant to a w +;29G11-Gal4DBD. In our experiments, Heterozygous flies were used. The codon-optimized Sensor cDNAs were used in the fruit fly. The flies were raised at a higher temperature of 21C.
Two-photon imaging took place under a ×40 0.8 NA water-immersion objective (Olympus) on a laser-scanning microscope (BrukerNano) with GaAsP photomultiplier tubes. Laser power at 920 nm was kept constant at 8 mW using a Pockels cell. There was no sign of lightening at this laser intensity. The emission dichroic was 580 nm and emission filters 511/20–25 nm. Images were 32 × 128 pixels with a frame rate at 372 Hz.
The visual stimulation was performed by using a digital micromirror device (DMD, LightCrafter), which was fired at 0.125hz onto a screen covering the visual field in front of the right eye. A blue LED (M470L3, Thorlabs) emitting through a 474/23–25-nm bandpass filter (to keep blue light from contaminating the green imaging channel) provided illumination.
Light coloring increased the amount of calcium in L2 cells. There was a measurement taken in the medulla layer 2. A target region image was chosen by testing each focal layer with 0.5-Hz full-field visual stimulation until a layer with maximum ΔF/F0 was identified. Within this layer there were 2–3 columns that produced a maximum response. A backgroundROI was chosen where nobody was visible, in addition to the region of interest containing the L2 columns. The mean background intensity was subtracted from the mean L2 ROI. Imaging then targeted this region over a protocol involving multiple tests, as shown in Supplementary Table 3.
We cross the flies with 20XUAS-IVS-Syn21-op1- GECI-p10 and 10XUAS-IVS-Myr tdTomato. The flies were used in the experiments. Sensor cDNAs were codon-optimized for Drosophila. The NMJ test is the same as in our previous study. In brief, female third instar larvae were dissected in chilled (4 °C) Schneider’s insect medium (Sigma) to fully expose the body wall muscles. The nerves were severed in a close proximity to the cord. Dissection medium was then replaced with room temperature HL-6 saline in which 2 mM CaCl2 and 7 mM L-glutamate is added to keep tetany in place. A mercury lamp (X-CITE exacte) light source was used for excitation, and out-of-objective power was kept less than 5 mW to reduce bleaching. Type Ib boutons on muscle 13 from segment A3–A5 were imaged while the corresponding hemi-segment nerve was stimulated with square voltage pulses (4 V, pulse width of 0.3 ms, duration of 2 s and frequency of 1–160 Hz) through a suction electrode driven by a customized stimulator. Bath temperature and pH were continuously monitored with a thermometer and pH metre, respectively, and recorded throughout the experiment. The filters for imaging were as follows: excitation of centre wavelength (CWL) = 472 nm and bandwidth (BW) = 30 nm, dichroic of 495 nm, and emission of CWL = 520 nm and BW = 35 nm. Images were captured with an EMCCD (Andor iXon 897) at 128.5 frames per second and acquired with Metamorph software. ROIs around boutons were manually drawn, and data were analysed with a custom Python script. The same value was calculated for each Discriminability index.
Variants were crossed to a pan-neuronal driver line, also containing tdTomato (pJFRC22-10XUAS-IVS-myr::tdTomato in su(Hw)attP8;; R57C10 at VK00020, R57C10 at VK00040). The third instar was fixed after being filleted. Primary chicken anti-GFP (1:1,000; A10262, Thermo Fisher) and secondary goat anti-chicken AlexaFluor Plus 488 (1:800; A32931, Thermo Fisher) were used to stain GECIs. Primary rabbit anti-RFP (1:1,000; 632496, Clontech) and secondary goat anti-rabbit Cy3 (1:1,000; 111-165-144, Jackson) labelled tdTomato.
The myr is co-expressed with the MB077B driver. Adults 3–6 days old were harvested, brains dissected and fixed using standard techniques. GCaMP variants were directly labelled with rabbit anti-GFP (1:500; AlexaFluor 488; A-21311, Molecular Probes). The goat anti-rat Cy3 (1,1,000; 112-235-181, Jackson) is labeled tdTomato.
ROIs were drawn on targeted regions using custom Python scripts. Within each ROI, otsu-thresholding was used to identify regions expressing myr::tdTomato. The intensity was taken for both the variant and tTomatod. The ratio is the intensity from the green channel (variant staining) divided by the intensity from the red channel (myr::tdTomato staining).
The same genes were used for the NMJ staining and the extract from female brains was made with them. Standard techniques were used to perform Westernblots. Each variant was stained using primary rabbit anti-GFP (PC408, Millipore Sigma) and secondary goat anti-rabbit IgG conjugated to horseradish peroxidase (HRP; 31460, Thermo Fisher/Invitrogen). Actin was stained using mouse IgM anti-α-actin (1:5,000; MA1-744, Thermo Fisher/Invitrogen) and goat anti-mouse IgG and IgM-HRP (1:5,000; 31430 and 62-6820, respectively, Thermo Fisher/Invitrogen). A signal was created using SuperSignal West Dura luminescence and then imaged with a Gel imager. The intensity was measured using a tool. Band intensity was divided from the variant to the actin band to determine the ratio.
Source: https://www.nature.com/articles/s41586-023-05828-9
Care of Laboratory Animals in the Janelia Research Campus (IACUC) and IBC committees: A pathogen-free laboratory animal environment for laboratory animals
All mice were cared for in compliance with the Guide for the Care and Use of Laboratory Animals. All experiments at Janelia were approved by the Janelia Research Campus IACUC and IBC committees. Janelia is accredited by theAAAALAC. Mice were maintained under specific pathogen-free conditions. There was a free standing, individually ventilated rack where mice were housed. The holding room was ventilated with 100% outside filtered air with 15–20 air changes hourly. Each cage had at least 8 g of bedding, and a red Mouse Tunnel, among other things. Mice were kept on a dark cycle. The temperature in the holding room was kept at a comfortable 72 F. Irradiated rodent laboratory chow (LabDiet 5053) was provided ad libitum.
Purkinje cell-specific expression is stimulated by co-injection of virus expressing Cre with a promoter fragment from the Purkinje cell. A 3-mm diameter circular coverslip glued to a donut-shaped 3.5-mm diameter coverslip (no. 1 thickness, Warner Instruments) was cemented to the craniotomy using dental cement (C&B Metabond, Parkell). A custom titanium head post was cemented to the skull.
Source: https://www.nature.com/articles/s41586-023-05828-9
Neuronal imaging using femtosecond-pulse lasers in microscopic media with an intramuscular sedate
Mice were kept on a warm blanket (37 °C) and anaesthetized using 0.5% isoflurane and sedated with chlorprothixene (20–30 μl at 0.33 mg ml−1, intramuscular). Imaging was performed with a custom-built two-photon microscope with a resonant scanner. The light source was an Insight femtosecond-pulse laser (Spectra-Physics) running at 940 nm. The objective was a 16 water immersed lens. The detection path consisted of a custom filter set (525/50 nm (functional channel), 600/60 nm (cell-targeting channel) and a 565-nm dichroic mirror) ending in a pair of GaAsP photomultiplier tubes (Hamamatsu). Images were acquired using ScanImage (vidriotechnologies.com)64. Functional images of L2/L3 cells were collected at a rate of around 30 or 122 degrees Centigrade. Unless otherwise stated, the laser power at the front of the objective was up to 50 mW.
The pipettes were filled with sterile saline. AlexaFluor 594. Recording of upper L2 neurons were obtained from the somatic cell attached recordings. Spikes can be recorded in either current or voltage mode. Signals were filtered at 20 kHz (Multiclamp 700B, Axon Instruments) and digitized at 50 kHz using Wavesurfer (wavesurfer.janelia.org/). The frames were recorded and offline synchronized with the recordings of the pulse of the brain. A randomized visual stimulation was delivered to increase the activity of the cells after establishment of a low resistance seal. We increased the firing probability of the recorded neuron by applying DC current in a small subset of recordings.
Visual stimuli were moving gratings generated using the Psychophysics Toolbox in MATLAB (Mathworks), presented using an LCD monitor (30 × 40 cm2), placed 25 cm in front of the centre of the right eye of the mouse. Each trial involved a 2s blank period and a 2-s drifting sinusoidal grating, with different frequencies and directions. ScanImage provides frame-start pulse which synchronized the stimuli to individual image frames.
Source: https://www.nature.com/articles/s41586-023-05828-9
Brain synaptic sensitivity and sensitivity measurements in loose-seal- recorded cell dendrite films using the Suite2p Python implementation
A mixture of ketamine-xylazine and 4% PFAS was given to the mice while they were in the loose-seal recording sessions. The brains were extracted and post-fixed overnight in the perfusing solution. The brains were sectioned at 50-m thickness. Triton X-100 (in PBS) for 1 h at room temperature, incubated with primary antibody (Rb-anti-GFP, 1:500; G10362, Invitrogen) for 2 days at 4 °C, and secondary antibody (AlexaFluor 594 conjugated goat anti-Rb, 1:500; A-11012, Invitrogen) overnight at 4 °C. The sections were mounted on microscope slides with Vectashield hard-set antifade mounting medium. The sample was imaged using the TissueFAXS 200 slide scanner and the X-Light V2 spinning disk confocalimaging system. Z2 microscope (Carl Zeiss Microscopy) equipped with a Plan-Apochromat ×20/0.8 M27 objective lens.
The movies were recorded in motion and categorized with the Suite2p Python implementation. The loose-seal- recorded cell’s ROI was manually selected from the automatically serpsed ones. For this dataset, we could calculate the neuropil contamination for most of the movies and got a distribution with a median of r_neu ~ 0.8 (Extended Data Fig. 14g–j), so we used this value uniformly for neuropil correction. The calcium events were defined by the grouping of APs. Then, we calculated F/F0 was the mean fluorescence value before the first AP in the group, where F is the instantaneous signal and F was defined separately for calcium events. The peak amplitudes were measured by taking into account the rise time of the calcium sensor relative to the last AP in the group, as well as the difference between the fluorescent intensity of the cell in the frame right before the first AP and it’s peak intensity after the 0–95% rise The index was used as a sensitivity.
Purkinje cell dendrite movies were captured without stimulation. The Python implementation of CaImAn69 was used to convert movies to F/F0 traces. Individual events within the traces were identified by the fact that they had the same local maximum in the F/F0 trace between them. Statistics for individual events were calculated by fitting the equation:
F’s are : 1: (1-E-(t-t_0)/ : tau _rmrise)
The rise and decay time constants are shaped by and Fmax is the maximum amplitude of the trace above Fstart.
A phenomenological model is used to convert spike times into a time series. This S2F model consists of two steps. At times spikes are converted to a variable called c(t) by using two double-exponential kernels.
Source: https://www.nature.com/articles/s41586-023-05828-9
Exact Results for the Goodness-of-fit of the S2F Model with Spikes and a Higher-Order Interquartile Range
There are two meanings in this: sum _t > t_k left.
Variance explained measures the goodness-of-fit of an S2F model, as (1-\frac{{(\Delta F/{F}{{\rm{raw}}}(t)-\Delta F/{F}{{\rm{Synth}}}(t))}^{2}}{{(\Delta Delta F/F_rmraw(t) > _t) Here we used only the time t with a spike rate of more than 0 Hz after spikes in the calculation, in which the instantaneous spike rate at time t is estimated by a boxcar-rolling average over a 600-ms time window.
Exact statistical tests used for each comparison, as well as n, are listed in the main text and figure legends. Box-and-whisker plots throughout the paper indicate the median and 25–75th percentile range; whiskers indicate the shorter of 1.5 times the interquartile range or the extreme data point71.
The Kruskal–Wallis test was used to find the P at a duration of 4 ms, and the pairwise comparison test was used to find jGCaMP7f. The Kruskal–Wallis test found P as high as 3.5 105. The pairwise comparison test to jGCaMP8f is as follows: jGCaMP8f P is 3.5 while jGCaMP8m is 2.8. The Kruskal–Wallis test found P at 3.4 10 5 at a time of 25 ms. The multiple comparison test for jGCaMP7f was conducted byDunn and the results were as follows. Numbers tested are the same as Extended Data Fig. 8c.
Source: https://www.nature.com/articles/s41586-023-05828-9
DNA synthesis, AAV particles and genetic studies of jGCaMP8s, jCGaMP8, f at Addgene and Bloomington Stock Center
DNA constructs and AAV particles of jGCaMP8s, jGCaMP8m and jGCaMP8f (pCMV, pAAV-synapsin-1, pAAV-synapsin-1-FLEX and pAAV-CAG-FLEX) have been deposited at Addgene (#162371–162382). The sequence has been deposited in the GenBank. jGCaMP8.80.80 has a crystal structure in the PDB. There are flies deposited at the Bloomington Stock Center and at Addgene. Please send the additional requests to [email protected].

